Protein Structure Alignment Methods and Approaches for Structural Analysis
Explore various methods such as geometric hashing, sequence-based alignment, and structural alignment approaches to measure alignment errors between closely related protein structures. Understand the significance of transformations, distance functions, and 3D rigid transformations in the alignment process.

Protein Structure Alignment Methods and Approaches for Structural Analysis
E N D
Presentation Transcript
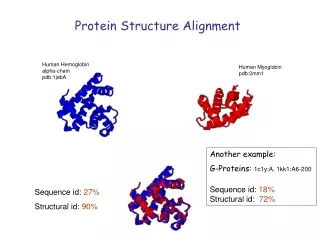
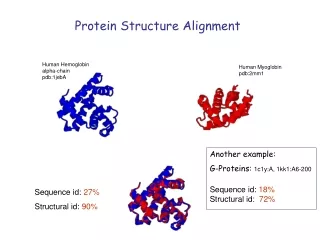
Sequence id: 27% Structural id: 90% Protein Structure Alignment Human Hemoglobin alpha-chain pdb:1jebA Human Myoglobin pdb:2mm1 Another example: G-Proteins: 1c1y:A, 1kk1:A6-200 Sequence id: 18% Structural id: 72%
Transformations • Translation • Translation and Rotation • Rigid Motion (Euclidian Trans.) • Translation, Rotation + Scaling
T Inexact Alignment. Simple case – two closely related proteins with the same number of amino acids. Question: how to measure an alignment error?
Distance Functions • Two point sets: A={ai} i=1…n • B={bj} j=1…m • Pairwise Correspondence: • (ak1,bt1) (ak2,bt2)… (akN,btN) (1) Exact Matching: ||aki – bti||=0 (2) Bottleneck max ||aki – bti|| (3) RMSD (Root Mean Square Distance) Sqrt( Σ||aki – bti||2/N)
Superposition - best least squares(RMSD – Root Mean Square Deviation) Given two sets of 3-D points : P={pi}, Q={qi} , i=1,…,n; rmsd(P,Q) = √ S i|pi - qi |2 /n Find a 3-D rigid transformation T* such that: rmsd( T*(P), Q ) = minT√ S i|T(pi) - qi |2 /n A closed form solution exists for this task. It can be computed in O(n) time.
T Correspondence is Unknown Given two configurations of points in the three dimensional space, find those rotations and translations of one of the point sets which produce “large” superimpositions of corresponding 3-D points.
A 3-D reference frame can be uniquely defined by the ordered vertices of a non-degenerate triangle p1 p2 p3
Sequence Based Structure Alignment • Run pairwise sequence alignment. • Based on sequence correspondence compute 3D transformation (least square fit can be applied). • Iteratively improve structural superposition. Not a good approach – sequence alignment can be incorrect.
Structure Alignment (Straightforward Algorithm) • For each pair of triplets, one from each molecule which define ‘almost’ congruent triangles compute the rigid transformation that superimposes them. • Count the number of aligned point pairs and sort the hypotheses by this number.
For the highest ranking hypotheses improve the transformation by replacing it by the best RMSD transformation for all the matching pairs. • Complexity : O(n3m3 ) * O(nm) . Applying 3D grid gives practically O(n3m3) * O(n) • If one exploits protein backbone geometry + 3D grid : O(nm) * O(n)
Structural Alignment Approaches Two interrelated problems: 3D transformation and point correspondence (matching, alignment) Some methods: • Generate a set of 3D transformations. • Cluster similar transformations. • Compute 3D alignment for each cluster representative. • Generate a set of 3D transformations. • Compute 3D alignment for each transformation. Geometric Hashing: Combines transformation and correspondence detection in one scheme.
i+k-1 i j+k-1 j Accuracy improvement during detection of 3D transformation. Instead of 3 points use more. How many? Align any possible pair of fragments - Fij(k)
Accept Fij(k) if rmsd(Fij(k)) <e. Complexity O(n3 n) * O(n)(assume n~m) (For each Fij(k) we need compute its rmsd) can be reduced to O(n3) * O(n)
k+l-1 k t+l-1 t Improvement : BLAST idea - detect short similar fragments, then extend as much as possible. i-1 i+1 i j-1 j+1 j ai-1aiai+1 bj-1 bjbj+1 Extend while: rmsd(Fij(k)) <e. Complexity: O(n2)*O(n)
4-helix bundle 2cbl:A 1f4n:A 1rhg:A 1b3q
Sequence Order Independent Alignment 2cbl:A 1f4n 1rhg:A 1b3q 51 103 113 169 chain A chain B 3 58 54 7 34 73 126 171 147 12 chain A chain B 306 355 354 305
The C2 domain calcium-binding motif • E. A. NALEFSKI and J. J. FALKE • The C2 domain calcium-binding motif: Structural and functional diversityProtein Sci 1996 5: 2375-2390
TRAF-Immunoglobulin Ensemble E- strand • Ensemble: 8 proteins from 2 folds. • Core: sandwich of 6 strands • Runtime: 21 seconds - helices ; - strands
Some Links • Rasmol – Molecular Visualization • SCOP - Structural Classification of Proteins • MultiProt -Protein Structural (pairwise/multiple) Alignment • MASS – Secondary Structure Based (pairwise/multiple) Alignment