Protein structure
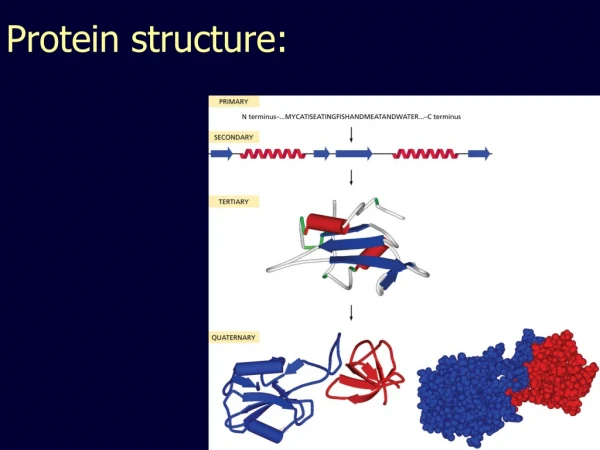
Protein structure. Primary structure Secondary structure Tertiary structure. Primary structure of polypeptides. The primary structure of a protein is specified by the nucleotide sequence of the corresponding gene.


Protein structure
E N D
Presentation Transcript
Protein structure • Primary structure • Secondary structure • Tertiary structure
Primary structure of polypeptides • The primary structure of a protein is specified by the nucleotide sequence of the corresponding gene. • Consist of amino acids linked together by amide bonds, forming a linear sequence of 100-1000 amino acids. • Contains all the information to confer both the three-dimensional structure of proteins in general and the catalytic activity of enzymes i particular.
Secondary structure found in proteins • Secondary structure is a term given to local regions (10-20 amino acids) of stable ordered three-dimensional structures held together by hydrogenbonding, that is non-covalent between acidic hydrogens (O-H, N-H) and lone pairs. • Some commonly observed secondary structure in proteins is :-helix, -sheet and -turn.
-helix • The -helix is a helical structure formed by a polypeptide chain in which hydrogen bonds are formed between the carbonyl oxygen of one amide linkage and N-H of the amide linkage four residues ahead in the chain.
-sheet • The -sheet is af structure formed by two or more linear polypeptide strands, held together by a series of interstrand hydrogen bonds. • There are two types of -sheet structures : parallel -sheets, in which the peptide strands both proceed in the same amino-to-carbibyl direction; and anti-parallel, in which the peptide strands proceed in opposite directions.
-turn • The -turn is often formed at the end of a -sheet which leads to a 180º turn in the direction of the polypeptide chain.
Tertiary structure of proteins • The three-dimensional structure of protein sub-units is known as the tertiary structure. It arises from packing together elements of secondary structure to form a stable global confirmation. • This involves burying hydrophobic amino acid side chain on the inside of the protein and positioning hydrophilic amino acid side chains on the surface. • Some common families of protein structure is : -helical, / structure and anti-parallel structures
-helical • The -helical proteins are made up of only -helices which is pack onto one another to form the tertiary sturcture. Structure of cytochrome b562
/ structure • The / structure consist of regular arrays of -sheet- -helix-parallel -sheet structures Structure of flavodoxin
Anti-parallel structures • The anti-parallel structures consist of regular arrays of -sheet- -turn-anti-parallel -sheet. Structure of suoeroxide dismutase
Watching proteins fold one molecule at a time The article provide evidence for a heterogeneous folding pathway (from primary structure to fully folded protein) for the protein adenylate kinase(adenylate kinase catalyse AMP+ATP⇌2ADP). Which is equal to a rugged energy landscape. It is predicted that a protein on such a surface may fold via a number of avaible pathways. Elizabeth Roades, Eugene Gussakovsky, and Gilad Haran
Method One of the major challenges to single-molecule protein-folding studies, is to restrict the molecule spatially such that temporal folding trajectories can be measured without modifying the conformtional dynamics of the protein.This problem is solves by a method where used the folding is followed, this involves trapping a single protein molecule (mutant C77A of AK from E.coli, where cystein residues were inserted at position 73 and 203) in a surface-tethered unilamellar lipid vesicles, with a diameter of 120nm. The AK molecule was labeled at position 73 (acceptor) and 203 (donor) by with fluorophore
Confirmation of unhindered movement of AK within the vesicles Before measuring the folding, its was confirm that AK molecules immobilized within lpid vesicles, were not interacting significantly with vesicular walls and thus able to freely diffuse and orient inside the vesicles. This was done by measuring the distribution o fluorescence polarization values of single vesicle-encapsulated AK molecules labeled with donor fluorophore only after excitation with circularly polarized light.A very narrow polarisation distribution was measured, which indicate a unhidered rotational motion of the molecules within the vescles. This is also confirmed by the absent of slow polarization changes.
FRET distribution of AK in vesicles under native and midtransition condition The conformational fluctuations of AK molecules, were studie by the use of fluorescence resonance energy transfer (FRET) between two specifically attched fluorophore. At first the EET distribution of vesicle-encapsulated labeled AK molecules were measured under native and denaturing conditions (2M Gdn·HCl, guanidide hydrochloride). The average FRET efficiency were 0,8 and 0,14 respectively, which are close to the ensemble values. This means that the encapsulation does not have an effect on the FRET values.
Because AK molecules are believe to deviate from a two-state folding, FRET distribution were also measured at 0,4-0,6M Gdn·HCl, which is around the midtransition conditions for AK 0,55M Gdn·HCl. Only the FRET distribution obtained from all trajectories taken at 0,4M Gdn·HCl is given in the article. This dstribution shows that the conformational space of the molecules can roughly be divide into two groups : the denatured ensemle ( EET< 0,45) and the folded ( EET > 0,45).
Examples of single molecule temporal trajectories There are some variability in the behavior of the trajectories. A molecule may move from one confirmation to another within the same ensemble (e.g B several transition within the folded ensemble) or transfer from one ensemble to the other (e.g D transition from the folded ensemble to the denatured ensemble).
Observation of Heterogeneous folding pathways After time-traces of individual vesicle-trapped AK molecules under midtransition condition, the EET trajectories were calculated. From these measurements a map of folding /unfolding transitions were obtained from singlemolecule trajectories. Each point represents the final vs. Initial FRET efficiency. Above the line are folding transition and below are unfolding transition. This shows a very large spread of the transitions. Which indicate strongly a large heterogenity of the folding reaction of AK.
A historam of the transition step size shows to peaks, one for folding and one for unfolding. The histogram also show that there is a preference for steps that change EET by 0,2-0,3. Thus AK molecules do not typically changes from fully folded to fully unfolded conformation and v.s in one step. This would require a change in EET 0,6. Instead AK molecules tend to jump through severeal intermediate steps. The exact sequence of intermediates changes from one molecule to another. Which indicate that AK molecules takes a different folding / unfolding pathway.
Slow transition Some trajectories show slow transitions either in the folding or unfolding direction, many of which take more than a second to complete. The slow gradual change in EET is a indication of a directed motion on the energy landscape, possible slow down by local traps. The directed motion can refelct a highly correlated non-markovian chain dynamics? or a result of slow growth of a folding nucleus?. Interestly, the slow transitions is not preceded by a fast jump, which means a abrupt change in EET , this suggest that the barriers sampled by these molecules are entropic rather then enthalpic.
Other possibilities for signal heterogeneity • Interaction of the partially denatured protein with the vesicle walls. This was ruled out by measurement of fluorescence polarization, which confirmed that the AK molecules were moving freely within the vesicles. • Jump of the fluorescent labels, thereby modifyng the FRET efficiency. This is not the case because NO FRET transitions are seen under native conditions, where orientational jumps should be just as abundant. • Occurence of cis-proline residues in the native state. Single proline isomerization cannot explaine the evidence of the large spread in folding transitions and small-step transitions.
Conclusion • AK molecules have a preference for small, partial folding/unfolding jumps, which indicate abundance of local traps on the free energy surface. • Occurence of slow, directed transitions, which involve correlated motions of protein segments