Structure Alignment
Structure Alignment. Structure Alignment. +. Content. Motivation Some basics Double Dynamic Programming. PART I: Motivation. Motivation: Conformational changes. Upon ligand binding structures may change Structural alignment can highlight the changes. GEFs. GAPs.


Structure Alignment
E N D
Presentation Transcript
Content • Motivation • Some basics • Double Dynamic Programming
Motivation: Conformational changes • Upon ligand binding structures may change • Structural alignment can highlight the changes
GEFs GAPs Conformational changes: Small GTPases • Small GTPases act as molecular switches to control and regulate important functions and pathways within in cell • Activated by guanine nucleotide exchange factors (GEF) • Inactivated by GTPase activating proteins (GAP)
G proteins: Conformational change in GTP and GDP bound state
Open and closed conformation of cytrate synthase (1cts,5cts) • Open: oxalacetate, Closed: oxalacetate and co-enzyme A • Loop between two helices moves by 6A and rotates by 28º, some atoms move by 10A
Hinge motion in Lactoferrin (1lfh, 1lfg) • Lactoferrin is an iron-binding protein found in secretions such as milk or tears • Rotation of 54º upon iron-binding
Hinge motion in Lactoferrin (1lfh, 1lfg) • Lactoferrin is an iron-binding protein found in secretions such as milk or tears • Rotation of 54º upon iron-binding
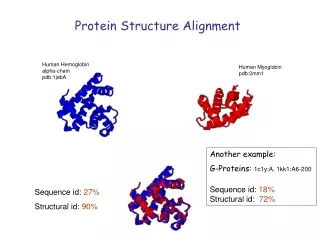
Motivation: (Distant) Relatives • Sequence similarity may be low, but structural similarity can still be high Picture from www.jenner.ac.uk/YBF/DanielleTalbot.ppt
Distant relatives • Globins occur widely • Primary function: binding oxygen • Assembly of helices surrounding haem group
Relatives Sperm whale myoglobin (2lh7) and Lupin leghaemoglobin (1mbd)
Relatives • Actinidin (2act) and Papain (9pap) • Sequence identity 49%, rmsd 0.77A • Same family: Papain-like
Relatives • Plastocyanin (5pcy) and azurin (2aza) • Core of structure is conserved
Relatives • Structure classifications like CATH and FSSP use structural alignments to identify superfamilies.
Sequence similarity: low >1cse Subtilisin AQTVPYGIPLIKADKVQAQGFKGANVKVAVLDTGIQA SHPDLNVVGGASFVAGEAYNTDGNGHGTHVAGTVAAL DNTTGVLGVAPSVSLYAVKVLNSSGSGSYSGIVSGIE WATTNGMDVINMSLGGASGSTAMKQAVDNAYARGVVV VAAAGNSGNSGSTNTIGYPAKYDSVIAVGAVDSNSNR ASFSSVGAELEVMAPGAGVYSTYPTNTYATLNGTSMA SPHVAGAAALILSKHPNLSASQVRNRLSSTATYLGSS FYYGKGLINVEAAAQ >1acb Chymotrypsin CGVPAIQPVLSGLSRIVNGEEAVPGSWPWQVSLQDKT GFHFCGGSLINENWVVTAAHCGVTTSDVVVAGEFDQG SSSEKIQKLKIAKVFKNSKYNSLTINNDITLLKLSTA ASFSQTVSAVCLPSASDDFAAGTTCVTTGWGLTRYTN ANTPDRLQQASLPLLSNTNCKKYWGTKIKDAMICAGA SGVSSCMGDSGGPLVCKKNGAWTLVGIVSWGSSTCST STPGVYARVTALVNWVQQTLAAN
Structural similarity: low 1CSE:E, 1ACB:E
Convergent Evolution • c.41.1 and b.47.1 share interaction partners d.40.1 CI-2 family of serine protease inhibitors d.58.3Protease propeptides/inhibitors c.41.1 Subtilisin-like b.47.1Trypsin-likeserine proteases d.84.1Subtilisin inhibitor c.56.5 Zn-dependentexopeptidase g.15.1 Ovomucoid/PCI-1 like inhibitor
Convergent Evolution 1oyv Ovomucoid/PCI-1 like inhibitor, g.15.1top Subtilisin like c.41.1bottom 1OYV 4sgb Ovomucoid/PCI-1 like inhibitor, g.15.1, top Trypsin-like serine proteases, b.47.1.2, bottom
Convergent Evolution • Aligned structures 1cse CI-2 family of serine proteases inhitors, d.40.1 top Subtilisin like c.41.1bottom 1acb CI-2 family of serine proteases inhitors, d.40.1 top Trypsin-like serine proteases, b.47.1.2, bottom
Catalytic Triad >1cse Subtilisin AQTVPYGIPLIKADKVQAQGFKGANVKVAVLDTGIQA SHPDLNVVGGASFVAGEAYNTDGNGHGTHVAGTVAAL DNTTGVLGVAPSVSLYAVKVLNSSGSGSYSGIVSGIE WATTNGMDVINMSLGGASGSTAMKQAVDNAYARGVVV VAAAGNSGNSGSTNTIGYPAKYDSVIAVGAVDSNSNR ASFSSVGAELEVMAPGAGVYSTYPTNTYATLNGTSMA SPHVAGAAALILSKHPNLSASQVRNRLSSTATYLGSS FYYGKGLINVEAAAQ >1acb Chymotrypsin CGVPAIQPVLSGLSRIVNGEEAVPGSWPWQVSLQDKT GFHFCGGSLINENWVVTAAHCGVTTSDVVVAGEFDQG SSSEKIQKLKIAKVFKNSKYNSLTINNDITLLKLSTA ASFSQTVSAVCLPSASDDFAAGTTCVTTGWGLTRYTN ANTPDRLQQASLPLLSNTNCKKYWGTKIKDAMICAGA SGVSSCMGDSGGPLVCKKNGAWTLVGIVSWGSSTCST STPGVYARVTALVNWVQQTLAAN
B C C Convergent evolution A and B are native, C is viral A B C A A’ Henschel et al., Bioinformatics 2006
HIV Nef mimics kinase in binding SH3 • Comparison of Nef-SH3 and intra-chain interaction of catalytic domain and SH3 of Hck, PDBs: 1efn and 2hck • No evidence of homology between Nef and Kinase Kinase (Src Haematopoeitic cell kinase, Catalytic domain) HIV1-Nef Fyn-SH3/Hck-SH3 Henschel et al., Bioinformatics 2006
Automatic calculation of equivalent residues Nef Kinase • Apart from PxxP motif matches: Arg71/Lys249, Phe90/His289 • Residues with equivalents are strictly conserved in HIV-Nef Henschel et al., Bioinformatics 2006
Mimickry of baculovirus p35 and human inhibitor of apoptosis • Caspase (red) • P35 (yellow) • IAP (green) • Upon infection cell starts apoptosis programme, p35 tries to stop it Henschel et al., Bioinformatics 2006
Mimickry of Capsids and Cyclophilin • HIV capsid protein (yellow) • Cyclophilin (red, green) • Cyclophilin A restricts HIV infectivity • Upon mutation of cyclophilin or inhibition with cyclophorin, infectivity goes up >100(Towers, Nature Medicine, 2003) Henschel et al., Bioinformatics 2006
What do we need? • To main operations to align structures: • Translation • Rotation • How to evaluate a structural alignment? • Root mean square deviation, rmsd
a b Root Mean Square Deviation • What is the distance between two points a with coordinates xa and ya and b with coordinates xb and yb? • Euclidean distance:d(a,b) = √(xa--xb )2 + (ya -yb )2 • And in 3D?
Root Mean Square Deviation • In a structure alignment the score measures how far the aligned atoms are from each other on average • Given the distances di between n aligned atoms, the root mean square deviation is defined as rmsd = √ 1/n ∑ di2
Quality of Alignment and Example • Unit of RMSD => e.g. Ångstroms • Identical structures => RMSD = “0” • Similar structures => RMSD is small (1 – 3 Å) • Distant structures => RMSD > 3 Å
A very simple algorithm… • …to align identical structures with conformational changes • Generate a sequence alignment (not necessary if both sequences are really 100% identical) • Compute center of mass for both structures • Move both structures so that the centers of mass are the origin • Compute the angle between all aligned residues • Rotate structure by median of all angles
A very simple algorithm… • …to align identical structures with conformational changes • Generate a sequence alignment (not necessary if both sequences are really 100% identical) • Compute center of mass for both structures • Move both structures so that the centers of mass are the origin • Compute the angle between all aligned residues • Rotate structure by median of all angles Question: How? Assume n atoms (x1,y1,z1) to (xn,yn,zn) (for one structure)
A very simple algorithm… Question: How?Assume n atoms(x1,y1,z1) to (xn,yn,zn:) Center of mass (xCoM,yCoM,zCoM) = (1/n ni=1 xi , 1/n ni=1 yi 1/n ni=1 zi ) • …to align identical structures with conformational changes • Generate a sequence alignment (not necessary if both sequences are really 100% identical) • Compute center of mass for both structures • Move both structures so that the centers of mass are the origin • Compute the angle between all aligned residues • Rotate structure by median of all angles Question: How?
A very simple algorithm… Question: How?Assume n atoms (x1,y1,z1) to (xn,yn,zn:) Center of mass (xCoM,yCoM,zCoM) = (1/n ni=1 xi , 1/n ni=1 yi 1/n ni=1 zi • …to align identical structures with conformational changes • Generate a sequence alignment (not necessary if both sequences are really 100% identical) • Compute center of mass for both structures • Move both structures so that the centers of mass are the origin • Compute the angle between all aligned residues • Rotate structure by median of all angles For all i: do xi:= xi-xCoM, yi:= yi-yCoM, yi:= yi-yCoM,
A very simple algorithm… • …to align identical structures with conformational changes • Generate a sequence alignment (not necessary if both sequences are really 100% identical) • Compute center of mass for both structures • Move both structures so that the centers of mass are the origin • Compute the angle between all aligned residues • Rotate structure by median of all angles Why median and not mean?
A refinement: Alternating alignment and superposition • 1. P = initial alignment (e.g. based on sequence alignment) • 2. Superpose structures A and B based on P • 3. Generate distance-based scoring matrix R from superposition • 4. Use dynamic programming to align A and B using scoring matrix R • 5. P‘ = new alignment derived from dynamic programming step • 6. If P‘ is different from P then go to step 2 again
Distance-based scoring matrix • Let d(Ai, Bj) be the Euclidean distance between Aiand Bj • Let t be the upper distance limit for residues to be rewarded • The scoring matrix R is defined as follows:R(Ai, Bj) = 1 / d(Ai, Bj) - 1 / t if R(Ai, Bj) > max. score then R(Ai, Bj) = max. score • The gap/mismatch penalty is set to 0
Distance-based scoring matrix • Let d(Ai, Bj) be the Euclidean distance between Aiand Bj • Let t be the upper distance limit for residues to be rewarded • The scoring matrix R is defined as follows:R(Ai, Bj) = 1 / d(Ai, Bj) - 1 / t if R(Ai, Bj) > max. score then R(Ai, Bj) = max. score • The gap/mismatch penalty is set to 0 What size doesPAM have? What size doesR have?
Example • R(Ai, Bj) = 1/d(Ai, Bj) - 1/t for t=1/10 and max. score =2