Protein Structure
900 likes | 1.33k Views
Protein Structure. IST 444. Protein Chemistry Basics. Proteins are polymers consisting of amino acids linked by peptide bonds Each amino acid consists of: a central carbon atom an amino group NH 2 a carboxyl group COOH a side chain (R group)

Protein Structure
E N D
Presentation Transcript
Protein Structure IST 444
Protein Chemistry Basics • Proteins are polymers consisting of amino acids linked by peptide bonds • Each amino acid consists of: • a central carbon atom • an amino group NH2 • a carboxyl group COOH • a side chain (R group) • Differences in side chains distinguish different amino acids.
repeating backbone structure repeating backbone structure CH2 CH2 CH CH2 H C CH3 CH2 CH2 CH2 CH2 COO- CH2 H3CCH3 CH2 HC CH CH2 CH2 CH3 HN N OH NH CH C NH2 N+H2 AspArgValTyrIleHisProPhe DRVYIHP F Protein sequence: DRVYIHPF O H O H O H O H O H O H O H H3N+ CH C N CH C N CH C N CH C N CH C N CH C N CH C N CH COO-
Side Chains Determine Structure • Hydrophobic stays inside, while hydrophilic stay close to water • Oppositely charged amino acids can form salt bridge. • Polar amino acids can participate hydrogen bonding
Steps in Obtaining Protein Structure Target selection Obtain, characterize protein Determine, refine, model the structure Deposit in repository
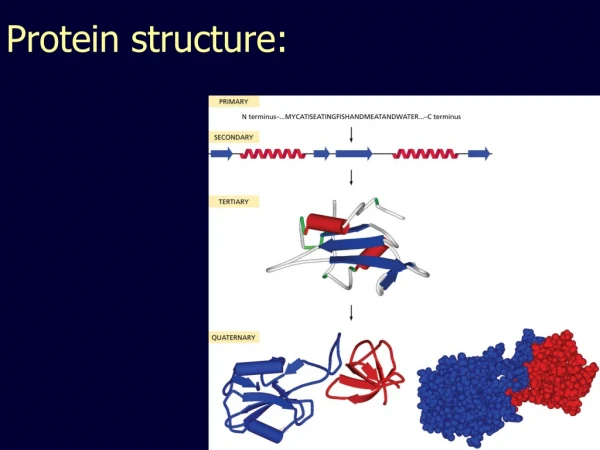
Domain, Fold, Motif • A protein chain could have several domains • A domain is a discrete portion of a protein, can fold independently, possess its own function • The overall shape of a domain is called a fold. There are only a few thousand possible folds. • Sequence motif: highly conserved protein subsequence • Structure motif: highly conserved substructure
Protein Data Bank Protein structures, solved using experimental techniques Unique structural folds Same structural folds Different structural folds
Protein Structure Determination • High-resolution structure determination • X-ray crystallography (~1Å) • Nuclear magnetic resonance (NMR) (~1-2.5Å) • Low-resolution structure determination • Cryo-EM (electron-microscropy) ~10-15Å
most accurate An extremely pure protein sample is needed. The protein sample must form crystals that are relatively large without flaws. Generally the biggest problem. Many proteins aren’t amenable to crystallization at all (i.e., proteins that do their work inside of a cell membrane). ~$100K per structure X-ray crystallography
Nuclear Magnetic Resonance • Fairly accurate • No need for crystals • limited to small, soluble proteins only.
Protein Structure Visualization • http://www.umass.edu/microbio/chime/top5.htm • http://molvis.sdsc.edu/visres/ • Rasmol • Chime • Protein Explorer • DeepView • JmolJava
Secondary Structure Prediction • Rules developed from PDB data • Chou and Fasman (1974) developed an algorithm based on the frequencies of amino acids found in a helices, b-sheets, and turns. • Proline: occurs at turns, but not in a helices. • http://prowl.rockefeller.edu/aainfo/chou.htm • Modern algorithms: use multiple sequence alignments and achieve higher success rate (about 70-75%)
Ramachandran Plot a way to visualize dihedral angles φ (phi) against ψ (psi) of amino acid residues in protein structure.
Chou Fasman 1974 • measured frequencies at which each amino acid appeared in particular types of secondary sequences in a set of proteins of known structure • assigns the amino acids three conformational parameters based on the frequency at which they were observed in alpha helices, beta sheets and beta turns • P(a) = propensity to form alpha helices • P(b) = propensity to form beta sheets • P(turn) = propensity to form beta turns • also assigns 4 turn parameters based on frequency at which they were observed in the first, second, third or fourth position of a beta turn • f(i) = probability of being in position 1 • f(i+1) = probability of being in position 2 • f(i+2) = probability of being in position 3 • f(i+3) = probability of being in position 4
Chou Fasman isn’t Perfect • Accuracy = 50-85%, depending on the protein • http://npsa-pbil.ibcp.fr/NPSA/npsa_references.html • Software and sites for protein predictions
GOR (Garnier, Osguthorpe and Robson) • Another commonly used algorithm, uses a window of 17 amino acids to predict secondary structure • rationale: experiments show each amino acid has a significant effect on the conformation of amino acids up to 8 positions in front or behind it. • a collection of 25 proteins of known structure was analyzed, and the frequency at which each amino acid was found in helix, sheet, turn or coil within the 17 position window was determined • this creates a 17 *20 scoring matrix that is used to calculate the most likely conformation of each amino acid within the 17 a.a. window • This window slides down the primary sequence, scoring the most likely conformation for each amino acid based on the neighboring amino acids. • Accuracy is about 65%
Signal for a Coiled Region • Gapped in multiple alignments • Small polar residues –Ala –Gly (v. small so flexible) –Ser –Thr • Prolines rarer in other kinds of secondary structure
Hidden Markov Models • Hidden Markov Models (HMMs) are a more sophisticated form of profile analysis. • Rather than build a table of amino acid frequencies at each position, they model the transition from one amino acid to the next. • Pfam is built with HMMs.
Discovery of new Motifs • All of the tools discussed so far rely on a database of existing domains/motifs • How to discover new motifs • Start with a set of related proteins • Make a multiple alignment • Build a pattern or profile
Depicting Structure Helix Beta Sheet Loop PDB ID: 12as
PDB New Fold Growth • Only a few thousand unique folds in nature • 90% of new structures deposited to PDB in the past three years have similar structural folds Old fold New fold
Secondary structure is context-dependent • Elements may be predicted to ID topology • Generally only 50% of a structure is alpha-helix or beta-sheet. • Beta-strands have necessarily longer range associations.
Secondary Structure • Protein secondary structure takes one of three forms: • Alpha helix • Beta pleated sheet • Turn • 2ndary structure is predicted within a small window • Many different algorithms, not highly accurate • Better predictions from a multiple alignment
Signals for Alpha Helices • Amphipathic helices interact with core and solvent • Characteristic hydrophobicity profile • Prolines disrupt the middles of helices
Signals for beta strands • Edge strands alternate hydrophobic/hydrophilic • Center strands all hydrophobic • Strands are extended so few residues per core span
Antiparallel Beta Sheet Parallel Beta Sheet Peptide chains have a directionality conferred by their N-terminus and C-terminus. β strands can be said to be directional, indicated by an arrow pointing toward the C-terminus. Adjacent β strands can form hydrogen bonds in antiparallel, parallel, or mixed arrangements. Antiparallel β strands alternate directions so that the N-terminus of one strand is adjacent to the C-terminus of the next. This produces the strongest inter-strand stability because it allows the inter-strand hydrogen bonds between carbonyls and amines to be planar, which is their preferred orientation.
R groups don’t form these secondary structures, but block formation of the secondary structures. The bonds forming the structures are from the amino and carboxy groups of the amino acid residues.
Creating Beta Sheets • Large aromatic residues (Tyr, Phe and Trp) and β-branched amino acids (Thr, Val, Ile) are favored to be found in β strands in the middle of β sheets. Interestingly, different types of residues (such as Pro) are likely to be found in the edge strands in β sheets
Protein Classification • Family: homologous, same ancestor, high sequence identity, similar structures • Super Family: distant homologous, same ancestor, sequence identity is around 25%-30%, similar structures. • Fold: only shapes are similar, no homologous relationship, low sequence identity. • Protein classification databases: Pfam, SCOP, CATH, FSSP
Pfam • http://www.sanger.ac.uk/Software/Pfam/ • Protein sequence classification database • As of Pfam 24.0 (October 2009, 11912 families) • Multiple sequence alignment for each family, then modeled by a HMM model
SCOP: Structural Classification of Proteins http://scop.mrc-lmb.cam.ac.uk/scop/ Protein structure classification database, manually curated 110800 Domains, 38221 PDB entries
SCOP • Nearly all proteins have structural similarities with other proteins and, in some of these cases, share a common evolutionary origin. • The SCOP database, created by manual inspection and automated methods, aims to provide a detailed and comprehensive description of the structural and evolutionary relationships between all proteins whose structure is known. • SCOP provides a broad survey of all known protein folds, detailed information about the close relatives of any particular protein, and a framework for future research and classification.
The Problem protein structure • Protein functions determined by 3D structures • ~ 30,000 protein structures in PDB (Protein Data Bank) • Experimental determination of protein structures time-consuming and expensive • Many protein sequences available medicine sequence function
Protein Structure Prediction • In theory, a protein structure can be solved computationally • A protein folds into a 3D structure to minimizes its free potential energy • The problem can be formulated as a search problem for minimum energy • the search space is enormous • the number of local minima increases exponentially Computationally it is an exceedingly difficult problem
Who Cares? • Long history: more than 30 years • Listed as a “grand challenge” problem • IBM’s big blue • Competitions: CASP (1992-2006) • Useful for • Drug design • Function annotation • Rational protein engineering • Target selection
Observations • Sequences determine structures • Proteins fold into minimum energy state. • Structures are more conserved than sequences. Two protein with 30% identity likely share the same fold.
What determines structures? • Hydrogen bonds: essential in stabilizing the basic secondary structures • Hydrophobic effects: strongest determinants of protein structures • Van der Waal Forces: stabilizing the hydrophobic cores • Electrostatic forces: oppositely charged side chains form salt bridges
Protein Structure Prediction • Stage 1: Backbone Prediction • Ab initio folding • Homology modeling • Protein threading • Stage 2: Loop Modeling • Stage 3: Side-Chain Packing • Stage 4: Structure Refinement The picture is adapted from http://www.cs.ucdavis.edu/~koehl/ProModel/fillgap.html
State of The Art • Ab inito folding (simulation-based method) 1998 Duan and Kollman 36 residues, 1000 ns, 256 processors, 2 months Do not find native structure • Template-based (or knowledge-based) methods • Homology modeling: sequence-sequence alignment, works if sequence identity > 25% • Protein threading: sequence-structure alignment, can go beyond the 25% limit
Common structural motifs Membrane spanning (GCG= TransMem) Signal peptide (GCG= SPScan) Coiled coil (GCG= CoilScan) Helix-turn-helix (GCG = HTHScan) “Super-secondary” Structure