Protein Structure Similarity
600 likes | 1.18k Views
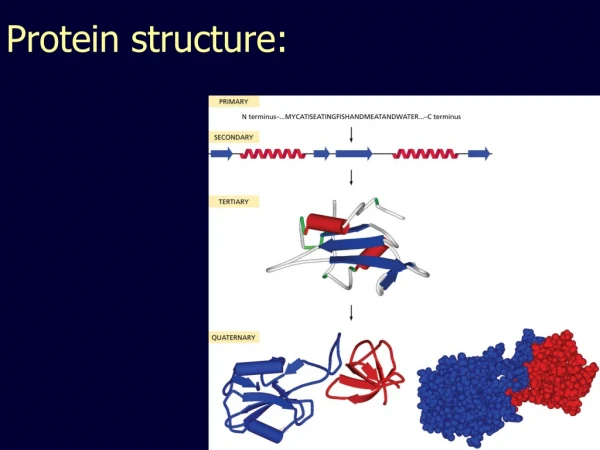
Protein Structure Similarity. Secondary Structure Elements: a helices , b strands/sheets , & loops. NMR spectrometry. Structure Prediction/Determination. Computational tools Homology, threading Molecular dynamics Experimental tools. X-ray crystallography.

Protein Structure Similarity
E N D
Presentation Transcript
Secondary Structure Elements: a helices, b strands/sheets, & loops
NMR spectrometry Structure Prediction/Determination • Computational tools • Homology, threading • Molecular dynamics • Experimental tools X-ray crystallography
Protein Structure Determination (1) • X-ray diffraction crystallography
Protein Structure Determination (2) • Nuclear magnetic resonance spectroscopy
Protein Data Bank 1990 250 new structures 1999 2500 new structures 2000 >20,000 structures total 2004 ~30,000 structures total
Protein Data Bank Only about 10% of structures have been determined for known protein sequences Protein Structure Initiative (PSI) 1990 250 new structures 1999 2500 new structures 2000 >20,000 structures total 2004 ~30,000 structures total
Structure Similarity • Refers to how well (or poorly) 3D folded structures of proteins can be aligned • Expected to reflect functional similarities (interaction with other molecules) Proteins in the TIM barrel fold family
Alignment of 1xis and 1nar (TIM-Barrels) ribbon format Sayle, R. RasMol. A protein visualization tool. http://www.umass.edu/microbio/rasmol/index2.htm. 1xis 1nar backbone format Alignment computed by DALI ahelix axes
Structure Similarity • Refers to how well (or poorly) 3D folded structures of proteins can be aligned • Is expected to reflect functional similarities (interaction with other molecules) • 2000: ~ 20,000 structures in PDB ~ 4,000 different folds (1:5 ratio)
Structure Similarity • Refers to how well (or poorly) 3D folded structures of proteins can be aligned • Is expected to reflect functional similarities (interaction with other molecules) • 2000: ~ 20,000 structures in PDB ~ 4,000 different folds (1:5 ratio) • Three possible reasons: - evolution, - physical constraints (e.g., few ways to maximize hydrophobic interactions), - limits in techniques used for structure determination • Given a new structure, the probability is high that it is similar to an existing one
sequencesimilarity Why Comparing Protein Folded Structures? • Low sequence similarity may yield very similar structures • Sometimes high sequence similarity yields different structures Sequence Structure Function
Alignment of 1xis and 1nar (TIM-Barrels) 1xis and 1nar have only 7% sequenceidentity, but approximately 70% of the residues are structurally similar
sequencesimilarity structuresimilarity Why Comparing Protein Folded Structures? • Low sequence similarity may yield very similar structures • Sometimes high sequence similarity yields different structures • Structure comparison is expected to provide more pertinent information about functional (dis-)similarity among proteins, especially with non-evolutionary relationships or non-detectable evolutionary relationships Sequence Structure Function
Ill-Posed Problem Multiple Terminology • (Dis-)similarity analysis • Structure comparison • Alignment, superposition, matching • Classification • Applications • Definitions and issues • Methods
A Few Web Sites • Protein Data Bank (PDB):http://www.rcsb.org/pdb/ • Protein classification: • SCOP:http://scop.berkeley.edu/ • CATHhttp://www.biochem.ucl.ac.uk/bsm/cath/ • Protein alignment: • DALI:http://www.ebi.ac.uk/dali/ • LOCK:http://motif.stanford.edu/lock2/
Application #1: Find Global Similarities Among Protein Structures • Given two protein structures, find the largest similar substructures • For example, a substructure is a subset of Ca atoms or a subset of secondary structure elements in each molecule • Several possible similarity measures • Variants: 1-to-1, 1-to-many, many-to-many (PDB) • Must be automatic (and fast)
Application #2: Classify Proteins • Many proteins, but relatively few distinct fold families [Chotia, 1992; Holm and Sander, 1996; Brenner et al. 1997] • Hierarchical classification • Insight into functions and structure stabilization • Basis for homology and threading • Manual classification SCOP [Murzin et al., 1995]
Application #2: Classify Proteins Class: Similar secondary structure content • Many proteins, but relatively few distinct fold families [Chotia, 1992; Holm and Sander, 1996; Brenner et al. 1997] • Hierarchical classification • Insight into functions and structure stabilization • Basis for homology and threading • Manual classification SCOP [Murzin et al., 1995] • Increasing size of PDB Automatic classifiers: CATH [Orengo et al., 1997]; Pclass [Singh et al.]; FSSP [Holm and Sander] Fold: SSE’s in similar arrangement Family: Clear evolutionary relationship
Application #3: Find Motif in Protein Structure • Given a protein structure and a motif (e.g., a small collection of atoms corresponding to a binding site) • Find whether the motif matches a substructure of the protein • Variant: One motif against many proteins Active sites of 1PIP and 5PAD. Only 3 amino-acids participate in the motif
Application #4: Find Pharmacophore • Given: • Small collection (5-10) of small flexible ligands with similar activity (hence, assumed to bind at same protein site) • Low-energy conformations (several dozens to few 100’s) for each ligand • Find substructure (pharmacophore) that occurs in at least one conformation of each ligand • Key problem in drug design when binding site is unknown
1TLP 4TMN 5TMN 6TMN The 4 ligands overlappedwith their pharmacophorematched Clusters of low-energy conformations of 1TLP Application #4: Find Pharmacophore Inhibitors of thermolysin
Application #5: Search for Ligands Containing a Pharmacophore • Given: • Database containing several 100,000, or more, small ligands • A pharmacophore P • Find all ligands that have a low-energy conformation containing P • Data mining of pharmaceutical databases (lead generation) S.M. LaValle, P.W. Finn, L.E. Kavraki, and J.C. Latombe. A Randomized Kinematics-Based Approach to Pharmacophore-Constrained Conformational Search and Database Screening. J. of Computational Chemistry, 21(9):731-747, July 2000
Applications • Definitions and issues • Methods
3D Molecular Structure • Collection of (possibly typed) atoms or groups of atoms in some given 3D relative placement • The placement of a group of atoms is defined by the position of a reference point (e.g., the center of an atom) and the orientation of a reference direction • The type can be the atom ID, the amino-acid ID, etc…
Matching of Structures Two structures A and B match iff: • Correspondence:There is a one-to-one map between their elements • Alignment:There exists a rigid-body transform T such that the RMSD between the elements in A and those in T(B) is less than some threshold e.
But a complete match is rarely possible: • The molecules have different sizes • Their shapes are only locally similar Alignment of 3adk and 1gky Both matching and non-matching secondary structure elements
Partial Match • Notion of support σ of the match: the match is between σ(A) and σ(B) • Dual problem: - What is the support? - What is the transform? • Often several (many) possible supports • Small supports motifs
Mathematical Relative g f s ||f - g||2 Over which support?
Mathematical Relative g f s ||f - g||2 Over which support?
A A σ(B) B B σ(A) Gap Distributed Support
A A B B What is Best? Should gaps be penalized?
A B What About This? Sequence along backbone is not preserved
Similarity measure is unlikely to satisfy triangular inequality for partial match
Scoring Issues • Trade-off between size of σ and RMSD • How should gaps be counted? • Is there a “quality” of the correspondence? [The correspondence may, or may not, satisfy type and/or backbone sequence preferences] • Should accessible surface be given more importance? • Similarity measure may be different from the inverse of RSMD (though no consensus on best measure!) • But RMSD is computationally very convenient!
Gap penalty Examples RMSD dissimilarity measure emphasizes differences smaller support STRUCTAL’s similarity measure emphasizes similarities larger support
Comparison of Similarity Measures A.C.M. May. Toward more meaningful hierarchical classification of amino acids scoring functions. Protein Engineering, 12:707-712, 1999reviews 37 protein structure similarity measures The difficulty of defining a similarity score is probably due to the facts that structure comparison is an ill-posed problem and has multiple solutions
Bottom Line Finding an optimal partial match is NP-hard: No fast algorithm is guaranteed to give an optimal answer for any given measure [Godzik, 1996] Heuristic/approximate algorithms Probably not a single solution, but application- dependent solutions But there exist general algorithmic principles
Computational Questions Given a (dis)similarity measure and two proteins, compute the best match: • Which support? • Which correspondence? • Which alignment transform?
Applications • Definitions and issues • Methods
Find Global Similarities Among Protein Structures • Input:Two sets of features (atoms or groups of atoms) {a1,…,an} and {b1,…,bm} belonging to two different proteins A and B • Output:- Maximal correspondence set C of pairs (ai,bj), where all ai and all bj are distinct- Alignment transform T such that the RMSD of the pairs (ai,T(bj)) is less than a given e • Several possible outputs Variant of the Largest Common Point Set problem[Akutsu and Halldorsson, 1994]
Possible Correspondence Constraints • Typed features:(ai,bj) is a possible correspondence pair iff Type(ai) = Type(bj) • Ordered features:(ai,bj) and (ai’,bj’), where i’>i, are possible correspondence pairs iff j’>j[E.g., sequence along backbone]
Some Existing Software Ca atoms: • DALI [Holm and Sander, 1993] • STRUCTAL [Gerstein and Levitt, 1996] • MINAREA [Falicov and Cohen, 1996] • CE [Shindyalov and Bourne, 1998] • ProtDex [Aung,Fu and Tan, 2003] Secondary structure elements and Ca atoms: • VAST [Gibrat et al., 1996] • LOCK [Singh and Brutlag, 1996] • 3dSEARCH [Singh and Brutlag, 1999]
RMSD ≠ Similarity But matches and RMSD’s are not exactly what we need In general, we need to computea similarity measure of the form maxT S(A,T(B))where S is more complex than RMSD Two-step approach: 1. Compute best matches using RMSD 2. Adjust transform to maximize similarity measure
Computation of Best Matches Two “simultaneous” subproblems • Find maximal correspondence set C • Find alignment transform T Chicken-and-egg issue: • Each subproblem is relatively simple: • If we knew C, we could compute T • If we knew T, we could get C by proximity • But the combination is hard !!!