Download

1 / 22
280 likes | 1.02k Views
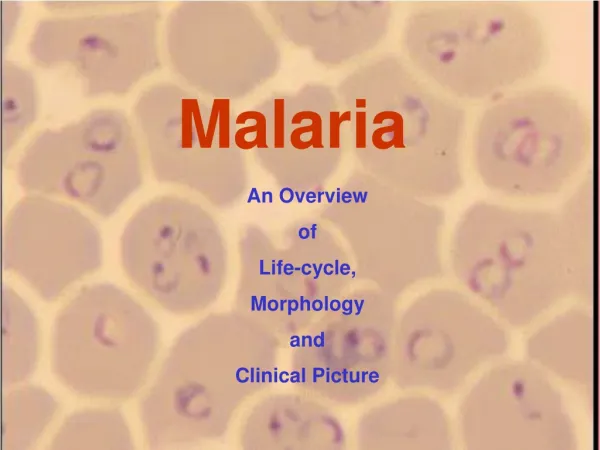
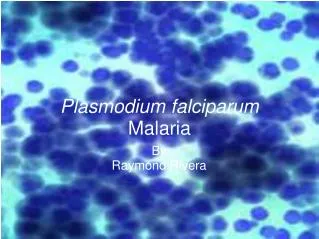
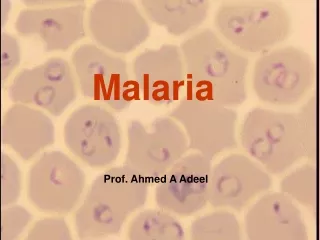
Malaria parasite (plasmodium). Pathogen of malaria: P.vivax ; P.falciparum ;P.malariae ; P.ovale _______________________________________ P.vivax ; P.falciparum are more common Plasmodium is a wide distribution in many tropical or subtropical regions of the world.

E N D
Malaria parasite (plasmodium) • Pathogen of malaria: • P.vivax ; P.falciparum ;P.malariae ; P.ovale • _______________________________________ • P.vivax ; P.falciparum are more common • Plasmodium is a wide distribution in many tropical or subtropical regions of the world
Characteristic of life cycle • Intermediate host : human • Final host : mosquito • Infective stage : sporozoite • Infective way : mosquito bite skin of human • Parasitic position : liver and red blood cells • Transmitted stage : gametocytes • Schizogonic cycle in red cells : 48 hrs/P.v • Sporozoite : tachysporozite and bradysporozite
Sporozoites, injected by Anopheles mosquitoes as they bite into the skin of mammalian hosts, rapidly enter the blood circulation to reach liver hepatocytes, where they mature in an entirely asymptomatic phase that lasts for approximately two weeks. Merozoites are liberated as merosomes from liver cells and then bud off from the hepatocytes to invade and develop in red blood cells (RBCs) (Sturm et al. 2006). In the RBCs, the parasites undergo asexual multiplication by schizogony and release merozoites, which invade other RBCs, thereby reinitiating the blood-stage cycle (Greenwood et al. 2008). The infected RBCs (iRBCs) are responsible for the disease symptoms, i.e., high and periodic fever (paroxysms), headaches (common to all human malaria species) and anaemia. The symptoms of P. falciparum include cerebral malaria and respiratory distress (life-threatening manifestations that are related to iRBC cytoadherence on microvascular endothelial cells), blockage of deep capillaries with neurological symptoms and death.
Life Cycle • sporozoites injected during mosquito feeding • invade liver cells • exoerythrocytic schizogony (merozoites) • merozoites invade RBCs • repeated erythrocytic schizogony cycles • gametocytes infective for mosquito • fusion of gametes in gut • sporogony on gut wall in hemocoel • sporozoites invade salivary glands
Exoerythrocytic Schizogony • hepatocyte invasion • asexual replication • 6-15 days • 1000-10,000 merozoites • no overt pathology
Erythrocytic Stage • intracellular parasite undergoes trophic phase • young trophozoite called ‘ring form’ • ingests host hemoglobin • cytostome • food vacuole • hemozoin (malarial pigment)
Invasive Stages • Merozoite • erythrocytes • Sporozoite • salivary glands • hepatocytes • Ookinete • epithelium
Clinical Features • characterized by acute febrile attacks (malaria paroxysms) • periodic episodes of fever alternating with symptom-free periods • manifestations and severity depend on species and host status • immunity, general health, nutritional state, genetics • recrudescences and relapses can occur over months or years • can develop severe complications (especially P. falciparum)
FARMACI ANTIMALARICI Three of the mainstays of malaria treatment come from natural products: quinine, lapachol (which led to atovaquone) and artemisinin.
GM: gametocytes; i.v.: intravenous injection; iRBC: infected red blood cells; LS: liver stages; MMV: Medicines for Malaria Venture
Chloroquine The discovery some 65 years ago of the exceptional antimalarial properties of chloroquine (CQ) rapidly paved the way for its massive use worldwide. After a decade of use, however, CQ resistance (CQR) emerged in a handful of origins, including in Southeast Asia, South America, and the Western Pacific region, from there spreading progressively throughout malaria-endemic areas including Africa where surges in malaria mortality were reported. Mechanism of action of chloroquine CQ, a weak base, can freely diffuse across membranes in its neutral form, and concentrates inside the acidic digestive vacuole (DV), preventing heme detoxification. CQ is thought to act by interfering with the digestion of haemoglobin in the blood stages of the malaria life cycle.
Azithromycin plus chloroquine: combination therapy for protection against malaria and sexually transmitted infections in pregnancy R Matthew Chico† & Daniel Chandramohan London School of Hygiene and Tropical Medicine, Faculty of Infectious and Tropical Diseases, Disease Control Department, London, UK Malaria infection in pregnancy is associated with low birth weight, preterm delivery, intrauterine growth-retardation and maternal anemia. Both azithromycin and chloroquine have been safely administered individually in all trimesters of pregnancy. The combination has demonstrated additive to synergistic effect in vitro and in vivo against Plasmodium falciparum. Azithromycin provide protection against several sexually transmitted and reproductive tract infections (STI/RTI) including Treponema pallidum, Neisseria gonorrhoeae, Chlamydia trachomatis, Trichomonas vaginalis, and possibly bacterial vaginosis as observed with other broad-spectrum antibiotics administered in the first half of pregnancy
Mechanisms of artemisinin drug action Mechanisms of artemisinin drug action, and the need for new antimalarial drugs effective against artemisinin-resistant P. falciparum. Considerable evidence suggests that the pharmacophoric peroxide bond in artemisinin undergoes reductive activation by heme released by hemoglobin digestion in the parasite digestive vacuole. This irreversible redox reaction produces carbon-centered radicals or carbocations that alkylate heme leading to oxidation reactions that damage parasite membranes. Indeed, the activity of artemisinin is dependent upon hemoglobin digestion, consistent with the specificity and efficacy of this drug against hemoglobin-degrading pathogens. Alternatively, it has been postulated that artemisinin oxidizes parasite FADH2 and parasite redox-active flavoenzymes or undergoes reductive activation in parasite mitochondria,both of which are thought to cause parasite death by an increase in reactive oxygen species.
Atovaquone is the end product of half a century of research by many groups who researched the antiparasitic properties of numerous structurally related compounds. Currently, atovaquone is used as a fixed-dose combination with proguanil (Malarone) for the treatment of children and adults with uncomplicated malaria or as a chemoprophylactic agent for preventing malaria in travellers. J Antimicrob Chemother 2013; 68: 977–985
Meccanismo di Azione Atovaquoneis a competitive inhibitor of ubiquinol, specifically inhibiting the mitochondrial electron transport chain at the bc1 complex (Coenzima Q). Inhibition of bc1 activity results in a loss of mitochondrial function. During the intra-erythrocytic stage of infection, a key role of the parasite mitochondrion is to provide orotate for pyrimidine biosynthesis through the activity of dihydroorotate dehydrogenase (DHODH). Consistent with this, inhibition of the bc1 complex by atovaquone affects the concentrations of metabolites in the pyrimidine biosynthetic pathway. Indeed, transgenic P. falciparum parasites expressing ubiquinone independent yeast DHODH have been shown to display an atovaquone-resistant phenotype. In addition, a recent study suggests that a further cellular consequence of mitochondrial inhibition by atovaquone is the inhibition of purine biosynthesis. Blood-stage parasite death as a result of atovaquone is relatively slow compared with other antimalarials such as artemisinin and chloroquine. This feature appears to be consistent with other mitochondrial-acting antimalarials and is possibly due to the drug acting only on late trophozoites and not on the earlier ‘ring’ stages. Atovaquone is, however, active against liver stages, resulting in its utility as a prophylactic drug; however, it is not believed to be active against ‘dormant’ hypnozoites.
Metabolism Under normal conditions, there is no evidence that atovaquone is significantly metabolized in humans, or that metabolism is required for drug elimination. Elimination Atovaquone pharmacokinetics are characterized by an extremely long elimination half-life of 50–84 h.59,63,65 Elimination is primarily via the liver, with almost undetectable amounts (,0.6%) of drug being eliminated via the kidneys.66 More than 90% of the drug excreted in bile was in the parent form. Elimination of atovaquone is complicated by the possibility of enterohepatic recirculation of the drug, which may help explain atovaquone pharmacokinetic profiles where a reduction and then an increase in drug concentration is seen with time.
Drug interactions Atovaquone is highly bound to plasma protein (.99.5%) and shows a high affinity for human serum albumin. Furthermore, the half-life of atovaquone is long, ranging from 50 to 84 h, and the major limiting factor to atovaquone clearance is probably its plasma protein binding. This suggests that any drug that reduces atovaquone plasma protein binding may potentially alter atovaquone tissue distribution and/or clearance. However, the authors can find no published articles investigating the drug-mediated displacement of atovaquone from plasma protein and the clinical impact of these interactions, and this area requires further research. The interaction observed between atovaquone and antiretrovirals, where efavirenz, lopinavir and ritonavir (all highly protein-bound drugs) reduced atovaquone plasma concentrations in HIV-infected patients, may involve atovaquone plasma protein displacement, although this was not demonstrated. This emphasizes the importance of establishing the interactions between antimalarials, including atovaquone, and antiretrovirals.
The global pipeline of new medicines for the control and elimination of malaria Melinda P Anthony, Jeremy N Burrows, Stephan Duparc, Joerg JMoehrle and Timothy NC Wells* • Changing priorities for the eradication agenda • Single-dose cure. • Transmission-blocking • Hypnozoite treatment and relapse prevention. • Chemoprevention (vaccinazione) Malaria Journal 2012, 11:316