Download

1 / 36
430 likes | 3.24k Views
Red cell Membrane Disorders Hereditary Spherocytosis . Hemolysis is defined as the premature destruction of red blood cells. Anemia results when the rate of destruction exeeds the capacity of the marrow to produce RBCs. Normal RBCs survival time is 110-120 days. Membrane Disorders

E N D
Hemolysis is defined as the premature destruction of red blood cells. Anemia results when the rate of destruction exeeds the capacity of the marrow to produce RBCs. Normal RBCs survival time is 110-120 days
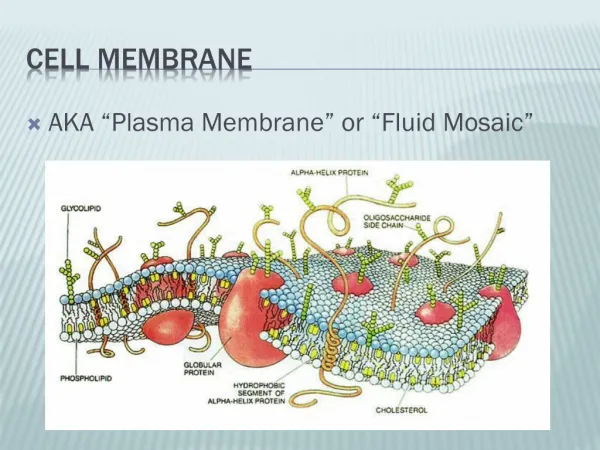
Membrane Disorders Inherited red cell membrane defects include : • Hereditary spherocytosis, • Hereditary elliptocytosis, • Hereditary stomatocytosis, and • Pyropoikilocytosis. In each abnormalities of the red cell membrane result in hemolysis.
What is Hereditary Spherocytosis? Spherocytosis, is an inherited disease that destroys red blood cells. This destruction of the red blood cells causes anemia. The shape of a normal red blood cell resembles a disk. Normal red blood cells easily change shape to move effectively through the small blood vessels between organs of the body. In spherocytosis the red blood cells are very round and have difficulty changing this shape. The lack of ability to change shapes makes moving through the small blood vessels difficult. Therefore, the red blood cells stay in the spleen longer than normal. This lengthy stay in the spleen damages the cell membranes.
HS is a common cause of hemolysis and hemolytic anemia. It is the most common inherited abnormality of the RBCs membrane. Prevalence: approximately 1/5000 people
Hereditary spherocytosis, the most frequent of the familial anemias, is inherited as an autosomal dominant trait; it is most common among people of northern European . The disease is most often identified during childhood or adolescence.
Pathophysiology Four abnormalities in red cell membrane proteins have been identified and include • spectrindeficiency alone, • combined spectrin and ankyrin deficiency, • (3) band 3 deficiency, and • (4) protein 4.2 defects. • Spectrindeficiency is the most common defect. Each is associated with a variety of mutations that result in different protein abnormalities and varied clinical expression.
A deficiency in spectrin, ankyrin,or protein 3 results in uncoupling in the vertical interactions of the lipid bilayer skeleton and the loss of membrane microvesicles. The loss of membrane surface area without a proportional loss of cell volume causes sphering of the RBCs.
Defects in vertical stabilization of the phospholipid bilayer of the RBC membrane cause separation of the spectrin - phospholipid bilayer. Portions of the phospholipid bilayer form vesicles and are lost from the RBC surface resulting in decreased surface area and spherocytosis.
Symptoms according to age Hydropsfetalis with death in utero can occur in the most severe cases . When detected in the neonatal period, HS is commonly accompanied by jaundice, requiring treatment with phototherapy or exchange transfusion .
Symptoms of Hereditary spherocytosis Anemia, jaundice, and splenomegaly are the clinical features of HS. Abnormal shaped red cells Anemia due to destruction of red blood cells Intermittent jaundice Enlarged spleen Biliary obstruction
Diagnosis of HS usually is established from: • 1.Family history • 2.Blood film • 3.Splenomegaly
Anemia, reticulocytosis, and spherocytosis on peripheral blood smear examination provide strong hints to suggest the diagnosis of HS.
Laboratory Studies The classic laboratory features of HS include: - Minimal anemia, -Reticulocytosis, -increased mean corpuscular hemoglobin concentration (MCHC), -Spherocytes on the peripheral blood smear, -hyperbilirubinemia, -Abnormal results on the Osmotic fragility test.
Differential diagnosis Other aquired causes of spherocytes in peripheral blood film: 1.Autoimmune hemolytic anaemias. 2.Hemolytic transfusion reactions. 3.Thermal injury
complications Gallstones Hemolytic crisis Aplastic crisis
Treatment : Splenectomyusually is curative Red cell survival is improved significantly but is not absolutely normal. The MCV usually falls, but the MCHC does not change significantly.
HEREDIATRY ELLIPTOCYTOSIS Definition: • Heterogeneous disorders with elliptical red cells. Types: • Common HE with discoidal elliptocytes. • Silent carriers :decrease expression of alpha spectrin • Hereditary pyropoikilocytosis • Spherocytic HE. • HF and HPP in neonates
mode of inheritance • The mode of inheritance is autosomal dominant, except for hereditary pyropoikilocytosis (HPP) which is autosomal recessive.
Physiology & Pathogenesis: • Mutations in or spectrin (commonest) leading to defective spectrin dimer formation. • Deficiency or dysfunction of protein 4.1. • Severity of haemolysis depends on the degree of spect def.
Clinical Features: • Variable, % of microcyte reflects severity. • Asymptomatic: carriers (parents or siblings of pts HE). • Transient haemolysis: Mild HE + infection, renal transplant rejection, B12 def., pregnancy.
Laboratory features: • Smear: diag. depends on % of elliptocytes (N < 5%) HE 25 – 90 % in +ve family history. • Elliptocytes: oval cells with long diameter > 2 times the short diameter. • Retics, LDH, bil., -ve DAT (Direct Antiglobulin Test) • OF: Mild HE normal OF. Severe HE OF. • Autohaemolysis N or (corrected by gl.).
SPHEROCYTIC ELLIPTOCYTOSIS Also called: HE with spherocytosis. Hereditary ovalocytosis. • Molecular basis unknown. Elliptical & spherocytes. Or round sphero-ovalocytes. • Clinically Haemolysis. • OF .
South –East Asian ovalocytosis Definition: Presence of oval elliptocytes and some with slit [RBC with a linear slit like central Pallor (N: circular)] genetics: autosomal dominant. Prevalence: Wide spread in Southeast Asia.
Characters: • Oval RBCs with: • 1 – 2 transverse ridges. • Or longitudinal slit. • Red cell rigidity. Clinical features: • Variable, jaundice at birth, pallor (variable), splenomegaly.
Laboratory features: • Anaemia. • Retics. • Smear: 10 – 25% stomatocytes. • OF & autohaemolysis. • Thermal stability. • Expression of certain red cell antigens. • Resistance to in vitro invasion by malaria