Download

1 / 21
210 likes | 331 Views
Metagene Projection addresses challenges in utilizing extensive array data due to variables like species, platform, and labeling methods. It claims to "reduce noise while still capturing the invariant biological features," allowing for improved clustering, class prediction, and sample contamination detection across platforms and species. The methodology uses Nonnegative Matrix Factorization (NMF) to effectively analyze gene expression profiles by focusing on metagenes. By leveraging model and test preprocessing parameter files, it enhances data quality and separation, leading to better analysis outcomes.

E N D
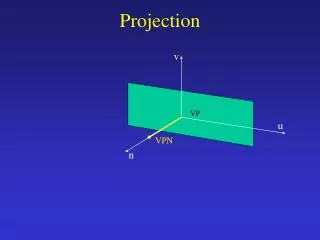
Metagene Projection • There are a lot of array data available • Species, platform, labeling method, researcher and other issues make using these data difficult. • Metagene Projection claims to “reduce noise while still capturing the invariant biological features of the data.” • This should “enable cross-platform and cross-species analysis, improve clustering and class prediction and provide a computational means to detect and remove sample contamination.”
NMF (Brunet et al., 2004 PNAS 101:4164) – Nonnegative Matrix Factorization W=genes X small # metagenes H= small # metagenes X samples M and T n (genes) x N (samples) KEY point: n (genes) identifiers in M and T must match Unknown: Can M and T be totally different types of data? Moore-Penrose generalized pseudoinverse
Model – 30 samples, 3 metagenes Test – 38 samples, 3 metagenes
After Metagene Projection Before Metagene Projection
Before Metagene Projection : Rank normalized and including only the top 500 markers of each class. – Underperforms metagene projection
KO of the same gene impacts different cell lines in similar way. • Both mouse stem cell lines, one on Exon array, one on 430_2
For 3’ UTR – max average per gene selected • For Exon – max probe count per transcript cluster id selected • gene symbol <–> gene symbol join • All 17354 genes used
Metagene Projection Preprocessing 2 required inputs for the genepattern metagene projection module are model and test preprocessing parameter files. gct.file="Arv.gct" cls.file="Arv.cls" column.subset="ALL" column.sel.type="samples" thres=3 ceil=14 fold=1 delta=1.5 norm=6 NO FILTER at this value 4525 pass
Model input, preprocessing and refinement H matrix from NMF
Projected data – possibly better separation move 1 het to improve clades