Clustering Gene Expression Data
Clustering Gene Expression Data. EMBnet: DNA Microarrays Workshop Mar. 4 – Mar. 8, 2002 ,UNIL & EPFL, Lausanne Gaddy Getz, Weizmann Institute, Israel. Gene Expression Data Clustering of Genes and Conditions Methods Agglomerative Hierarchical: Average Linkage Centroids: K-Means


Clustering Gene Expression Data
E N D
Presentation Transcript
Clustering Gene Expression Data EMBnet: DNA Microarrays Workshop Mar. 4 – Mar. 8, 2002 ,UNIL & EPFL, Lausanne Gaddy Getz, Weizmann Institute, Israel • Gene Expression Data • Clustering of Genes and Conditions • Methods • Agglomerative Hierarchical: Average Linkage • Centroids: K-Means • Physically motivated: Super-Paramagnetic Clustering • Coupled Two-Way Clustering
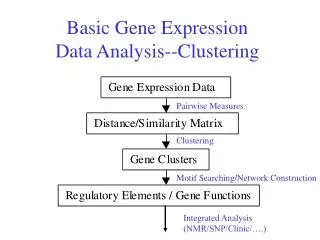
Gene Expression Technologies • DNA Chips (Affymetrix) and MicroArrays can measure mRNA concentration of thousands of genes simultaneously • General scheme: Extract RNA, synthesize labeled cDNA, Hybridize with DNA on chip.
Single Experiment • After hybridization • Scan the Chip and obtain an image file • Image Analysis (find spots, measure signal and noise)Tools: ScanAlyze, Affymetrix, … • Output File • Affymetrix chips: For each gene a reading proportional to the concentrations and a present/absent call.(Average Difference, Absent Call) • cDNA MicroArrays: competing hybridization of target and control. For each gene the log ratio of target and control. (CH1I-CH1B, CH2I-CH2B)
Preprocessing: From one experiment to many • Chip and Channel Normalization • Aim: bring readings of all experiments to be on the same scale • Cause: different RNA amounts, labeling efficiency and image acquisition parameters • Method: Multiply readings of each array/channel by a scaling factor such that: • The sum of the scaled readings will be the same for all arrays • Find scaling factor by a linear fit of the highly expressed genes • Note: In multi-channel experiments normalize each channel separately.
Preprocessing: From one experiment to many • Filtering of Genes • Remove genes that are absent in most experiments • Remove genes that are constant in all experiments • Remove genes with low readings which are not reliable.
Noise and Repeats log – log plot • >90% 2 to 3 fold • Multiplicative noise • Repeat experiments • Log scaledist(4,2)=dist(2,1)
We can ask many questions? Supervised Methods(use predefined labels) • Which genes are expressed differently in two known types of conditions? • What is the minimal set of genes needed to distinguish one type of conditions from the others? • Which genes behave similarly in the experiments? • How many different types of conditions are there? Unsupervised Methods(use only the data)
Unsupervised Analysis • Goal A:Find groups of genes that have correlated expression profiles.These genes are believed to belong to the same biological process and/or are co-regulated. • Goal B:Divide conditions to groups with similar gene expression profiles.Example: divide drugs according to their effect on gene expression. Clustering Methods
Cluster Analysis Yields Dendrogram T (RESOLUTION)
What is clustering? More Mathematically • Input: N data points, Xi, i=1,2,…,N in a D dimensional space. • Goal: Find “natural” groups or clusters. Data point of same cluster - “more similar” • Tasks: • Determine number of clusters • Generate a dendrogram • Identify significant “stable” clusters
Clustering is ill-posed • Problem specific definitions • Similarity: which points should be considered close? • Correlation coefficient • Euclidean distance • Resolution: specify/hierarchical results • Shape of clusters: general, spherical.
Similarity Measure • Similarity measures • Centered Correlation • Uncentered Correlation • Absolute correlation • Euclidean
2 4 5 3 1 1 3 2 4 5 Need to define the distance between thenew cluster and the other clusters. Single Linkage: distance between closest pair. Complete Linkage: distance between farthest pair. Average Linkage: average distance between all pairs or distance between cluster centers Agglomerative Hierarchical Clustering Distance between joined clusters The dendrogram induces a linear ordering of the data points Dendrogram
Agglomerative Hierarchical Clustering • Results depend on distance update method • Single Linkage: elongated clusters • Complete Linkage: sphere-like clusters • Greedy iterative process • NOT robust against noise • No inherent measure to choose the clusters
Centroid Methods - K-means • Start with random position of K centroids. • Iteratre until centroids are stable • Assign points to centroids • Move centroids to centerof assign points Iteration = 0
Centroid Methods - K-means • Start with random position of K centroids. • Iteratre until centroids are stable • Assign points to centroids • Move centroids to centerof assign points Iteration = 1
Centroid Methods - K-means • Start with random position of K centroids. • Iteratre until centroids are stable • Assign points to centroids • Move centroids to centerof assign points Iteration = 1
Centroid Methods - K-means • Start with random position of K centroids. • Iteratre until centroids are stable • Assign points to centroids • Move centroids to centerof assign points Iteration = 3
Centroid Methods - K-means • Result depends on initial centroids’ position • Fast algorithm: compute distances from data points to centroids • No way to choose K. • Example: 3 clusters / K=2, 3, 4 • Breaks long clusters
Super-Paramagnetic Clustering (SPC)M.Blatt, S.Weisman and E.Domany (1996) Neural Computation • The idea behind SPC is based on the physical properties dilute magnets. • Calculating correlation between magnet orientations atdifferent temperatures (T). T=Low
Super-Paramagnetic Clustering (SPC)M.Blatt, S.Weisman and E.Domany (1996) Neural Computation • The idea behind SPC is based on the physical properties dilute magnets. • Calculating correlation between magnet orientations atdifferent temperatures (T). T=High
Super-Paramagnetic Clustering (SPC)M.Blatt, S.Weisman and E.Domany (1996) Neural Computation • The idea behind SPC is based on the physical properties dilute magnets. • Calculating correlation between magnet orientations atdifferent temperatures (T). T=Intermediate
Super-Paramagnetic Clustering (SPC) • The algorithm simulates the magnets behavior at a range of temperatures and calculates their correlation • The temperature (T) controls the resolution • Example: N=4800 points in D=2
Output of SPC A function (T) that peaks when stable clusters break Size of largest clusters as function of T Dendrogram Stable clusters “live” for large T
Advantages of SPC • Scans all resolutions (T) • Robust against noise and initialization -calculates collective correlations. • Identifies “natural” () and stable clusters (T) • No need to pre-specify number of clusters • Clusters can be any shape
Many clustering methods applied to expression data • Agglomerative Hierarchical • Average Linkage (Eisen et. al., PNAS 1998) • Centroid (representative) • K-Means (Golub et. al., Science 1999) • Self Organized Maps (Tamayo et. al., PNAS 1999) • Physically motivated • Deterministic Annealing (Alon et. al., PNAS 1999) • Super-Paramagnetic Clustering (Getz et. al., Physica A 2000)
Available Tools • Software packages: • M. Eisen’s programs for clustering and display of results (Cluster, TreeView) • Predefined set of normalizations and filtering • Agglomerative, K-means, 1D SOM • Web sites: • Coupled Two-Way Clustering (CTWC) websitehttp://ctwc.weizmann.ac.il both CTWC and SPC • http://ep.ebi.ac.uk/EP/EPCLUST/ • General mathematical tools • MATLAB • Agglomerative, public m-files. • Statistical programs (SPSS, SAS, S-plus)
Back to gene expression data • 2 Goals: Cluster Genes and Conditions • 2 independent clustering: • Genes represented as vectors of expression in all conditions • Conditions are represented as vectors of expression of all genes
First clustering - Experiments 1. Identify tissue classes (tumor/normal)
Second Clustering - Genes Ribosomal proteins Cytochrome C metabolism HLA2 2.Find Differentiating And Correlated Genes
Coupled Two-Way Clustering (CTWC)G. Getz, E. Levine and E. Domany (2000) PNAS • Motivation: Only a small subset of genes play a role in a particular biological process; the other genes introduce noise, which may mask the signal of the important players. Only a subset of the samples exhibit the expression patterns of interest. • New Goal: Use subsets of genes to study subsets of samples (and vice versa) • A non-trivial task – exponential number of subsets. • CTWC is a heuristic to solve this problem.
Football Booing Cheering
CTWC of colon cancer data Tumor Normal (A) Protocol A Protocol B (B)
Glioma cell line Low grade astrocytoma Secondary GBM Primary GBM p53 mutation CTWC of Glioblastoma Data – S1(G5) Godard, Getz, Kobayashi, Nozaki, Diserens, Hamon, Stupp, Janzer, Bucher, de Tribolet, Domany & Hegi (2002) Submitted S14 S13 S12 S11 S10 AB004904 STAT-induced STAT inhibitor 3 M32977 VEGFANGIOGENESIS M35410 IGFBP2 X51602 VEGFR1ANGIOGENESIS M96322 Gravin AB004903 STAT-induced STAT inhibitor 2 X52946 PTN J04111 C-JUN X79067 TIS11B
Biological Work • Literature search for the genes • Genomics: search for common regulatory signal upstream of the genes • Proteomics: infer functions. • Design next experiment – get more data to validate result. • Find what is in common with sets of experiments/conditions.
Summary • Clustering methods are used to • find genes from the same biological process • group the experiments to similar conditions • Different clustering methods can give different results. The physically motivated ones are more robust. • Focusing on subsets of the genes and conditions can uncover structure that is masked when using all genes and conditions http://ctwc.weizmann.ac.il