Download
1 / 62
750 likes | 1.26k Views
Genetics of Mitochondrial Respiratory Chain Disorders. nucleus. mitochondria. Dept Medical Genetics Zhong-Shang University, China May 28, 2007. Lee-Jun C. Wong, Ph.D. Molecular and Human Genetics Baylor College of Medicine. mitochondrion.

E N D
Genetics of Mitochondrial Respiratory Chain Disorders nucleus mitochondria Dept Medical Genetics Zhong-Shang University, China May 28, 2007 Lee-Jun C. Wong, Ph.D. Molecular and Human Genetics Baylor College of Medicine
mitochondrion The only animal cellular organelle that contains its own DNA Hundreds to thousands of mitochondria per cell Egg cells: ~200,000, sperm cells: ~10 2-10 copies of mtDNA per mitochondrion
NAD NADH Proton gradient FAD FADH2 O2 H2O ADP ATP Major Function of mitochondria: electron transport chain Oxidative Phosphorylation producing energy, ATP Outer membrane Matrix Inner membrane Electron transport chain
Respiratory chain subunits encoded by two genomes: Nuclear and Mitochondria I:NADH DH II: SDH III: cyt c red IV: COX V: ATPase complex Mito/ nuclear 7/43 0/4 1/11 3/13 2/13
cytosol Outer membrane Inner membrane Matrix
Unique Features of Mitochondrial Genome • No introns • Except ~1.2kb (D-loop) at the origin of replication • the remaining are coding regions • Both strands are transcribed • ND6 is encoded by light strand • ATP6 and ATP8 are overlapped using • different reading frame • Mutations have been reported in all 13 mRNA, • 2rRNA, and all tRNA (except tRNAArg) • Polycistronic
Characteristics of Mitochondrial Genetics • Maternal inheritance • High Mutation Rate • limited proof reading & repair • Lack of protective histone proteins • close to the site of ROS production • Heteroplasmy • Threshold Effect • Heterogeneous Expression • Mitotic segregation
Homoplasmy 0 or 100% Heteroplasmy Between 0-100% Homoplasmy and Heteroplasmy
Mitochondrial DNA : common point mutations • MELAS: Mitochondrial Encephalopathy Lactic Acidosis and Stroke-like episodes. A3243G (80%), T3271C, in tRNALeu(UUR) • MERRF: Myoclonic epilepsy, Ragged Red Fibers. A8344G (80%), T8356C, in tRNALys • NARP: Neuropathy, Ataxia, Retinitis Pigmentosa. Leigh disease. T8993G, T8993C, in ATPase 6 • LHON: Leber Hereditary Optic Neuropathy. G11778A, G3460A, in ND4 and ND1 • Diabetes/deafness: A3243G
MELAS: Mitochodrial Encephalopathy Lactic Acidosis and Stroke-like episodes • The most common mtDNA point mutation: A3243G (80%) • A severe mutation, usually heteroplasmy. Homoplasmy not seen • Disease severity correlates with levels of mutant loads in affected tissues • Sporadic or maternal inheritance • Mechanism of pathogenesis • Abn RNA processing • tRNA post translation modification • Stability of tRNA • Aminoacylation • Protein translation
Mutation hot spot
MERRF: Myoclonic Epilepsy, Ragged Red Fibers • Most common merrf mutation is A8344G (80%) • usually heteroplasmy, not as severe as A3243G mutation, higher threshold • Disease severity correlates with levels of mutant loads in affected tissues • Require high level of mutant load (>60%) to show clinical symptoms • Mitochondrial proliferation
NARP: N = Neuropathy/Neurogenic weakness A = Ataxia RP = Retinitis pigmentosa MRNA mutationsT8993G (Leu to Arg in ATPase6): Continuous phenotypic spectrumnl > RP > NARP > Leigh syndromedepends on % mutant heteroplasmy
Leigh Syndrome • Mitochondrial encephalopathy • Presents in infancy • Psychomotor regression • Signs of brainstem dysfunction • Ataxia • Often fatal • Characteristic MRI findings
0% Spectrum of Clinical Phenotypes for T8993GBased on Percentage of Mutant Mitochondria “ Normal” 60% Retinitis Pigmentosa 75% NARP 90% Carelli et al. (2002) Arch Neurol 59: 264-270. Leigh Syndrome 100%
Percentage of mtDNA in Leucocytes Carrying the T8993G Mutation 31% carrier 80% NARP 94% Leigh 82% Phenotypically normal Failure to thrive Developmental delay hypotonia Failure to thrive Developmental regression Hypotonia Seizures Abn MRI
T8993G NARP/Leigh syndrome: a continuous phenotypical spectrum • Roughly correlates with heteroplasmy • Heteroplasmy variation important • Known heteroplasmy may not fully explain all the variation in phenotype • Prenatal testing: caution • Age • Tissue distribution • Modifier gene • Genetic background
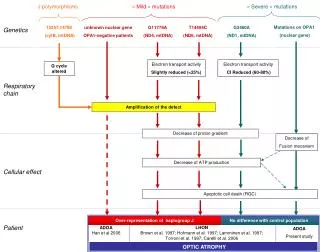
Leber’s Hereditary Optic Neuropathy (LHON) • Mostly involve homoplasmic mutations: 80% G11778A in ND4, 15% G3460A in ND1 • A degenerative eye disease • Age of onset: mid 20’s • Variable penetrance, 20-80%, with male to female ratio of about 4 to 1 • Missense mutation in conserved domain of complex 1 subunits • Primary mutations and secondary mutations
I d86y II d48y 1 2 3 5 6 7 8 4 III d2y d15y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 IV G14459A mutation A72V in ND6 Variable expression: LHON, dystonia III-6 NF1 unaffected IV-8 Hemiparesis NF1 Global delay Dysarthria spasticity MRS lactate peak IV-10 Stroke Dystonia Developmental delay short Spasticity Hearing loss MRS lactate peak IV-2 Limp Hemiparesis MRS lactate peak IV-9: unaffected All Homoplasmy
Proband, patient IV-10: Bilateral increased T2 signal in the putamen MRS shows elevated lactate Gropman, chen, Perng, Krasnewich, Chernoff, Tifft, and Wong. AJMG 2004;124A:377-382
patient IV-2 patient IV-8 bilateral symmertric increased T2 signal in the putamen unilateral increased T2 signal in the putamen Gropman, chen, Perng, Krasnewich, Chernoff, Tifft, and Wong. AJMG 2004;124A:377-382
mtDNA point mutations • tRNA • pathogenic ones are usually heteroplasmic • Affect overall mito protein translation, all subunits encoded by mtDNA • mRNA • Affect a specific protein subunit • Homoplasmic missense mutations do occur • Distinguish primary mutations and secondary mutations
A3243G Melas family M: 90%
A3243G: diabetes, hearing loss, retinopathy 48 y o 47 y o B: 12% H: 33% C: 30% B: 8% H: 6% C: 18% Diabetes Hypertension Heart disease Diabetes Hearing loss Macular pattern retinal dystrophy 28 y o B: 23% H: 15% C: 16% asymptomatic Am J Ophthalmol 1997;124:219
A8344G MERRF family I 1 2 3 4 5 II B: 0% 16% 14% 1 2 3 6 7 8 4 5 1993, B:48% 1995, B:62% 1995, H:60% III ~0% ~0% 0% ~0% B: 4% 30% 9 10 11 12 1 2 3 4 5 6 7 8 13 14 15 16 IV 1993, B:75% 65% 43% 1995, B:nd 65% 54% 1995, H:nd 65% 43% B: 5% ~0% ~0% 10% ~0% ~0% ~0% ~0% ~0% 5%
Mitochondrial Cardiomyopathy and peripheral neuropathy Mutation in tRNA lys (8363G>A) 1 2 3 4 I 76% 6 8 7 9 10 5 2 3 4 1 II 73% 88% 82% 60% 83% 4 5 1 2 3 6 III 94% 90% 84% 98% 91% 87% 73%
Single deletion Multiple deletion depletion
F16498-R32 mtDNA deletions F3212-R3319 F12093-R12170 F8389-R8529
Mitochondrial DNA Deletion Syndrome • Kearns Sayre syndrome • Ophthalmoplegia (inability to move eyes) • Ptosis (droopy eyes • Onset second decade • muscle • Pearson syndrome • Sideroblastic anemia with pancytopenia • Exocrine pancreatic insufficiency • Onset: early infancy • Blood • Multisystemic disease • PEO • Mitochondrial myopathy
Muscle or Blood? KSS vs Multisystemic Disorder
6 yo boy presented with Addison disease, Died of ARDS at 8 years of age Deletion mutant in Autopsy tissues 5 kb common deletion in every autopsy tissue
1 2 I heart problems 2 1 II 4 5 3 1 2 III 39 39 34 23 wheelchair bound MR cleft lip 1 IV 2 3 14 Clearly Kearns Sayre Syndrome, but deletion was not detected in blood.
Disorders of intergenomic signaling mtDNA multiple deletion anddepletion syndrome • Caused by nuclear genes responsible for the maintenance of mtDNA integrity, genes involved in mtDNA replication and balance of dNTP pools
MPV17 DNC DNA replication Transcription Translation Spinazzola and Zeviani, Gene 354 (2005) 162-168
DNA polymerase gamma mutations • Cause mtDNA multiple deletions and depletion • Autosomal recessive: eg, Alpers synd (infantile CNS and liver disease) • Autosomal dominant: Progressive external ophthalmoplegia
Autosomal dominant form of progressive external ophthalmoplegia (adPEO) • Twinkle gene: DNA helicase • ANT1 (Adenine Nucleotide Translocase 1) • POLG
Hepatocerebral form of mtDNA depletion syndromeinfantile hepatic failure • DGUOK (deoxyguanosine kinase) • MPV17, a mitochondrial inner membrane protein • POLG Autosomal recessive
DGUOK mutations cause mtDNA depletion and respiratory chain enzyme deficiencies Hepatocerebral form of mtDNA depletion syndrome P.W65X + c.487ins4 + P.W65X C.487ins4 P.W65X C.487ins4 Both mutations are deleterious. Missense mutations in DGK appear to have similar clinical phenotype
The liver biopsy showed portal fibrosis with extension into the lobule to surround hepatocytes. The hepatocytes are large with microvesicular steatosis and oncocytic change. The liver biopsy showed portal fibrosis with extension into the lobule to surround hepatocytes. The hepatocytes are large with microvesicular steatosis and oncocytic change.
Myopathic form of mtDNA depletion syndrome • TK2 (thymidine kinase) MNGIE Mitochondrial NeuroGastroIntestinal Encephlomyopathy • TP (thymidine phosphorylase) Both are Autosomal Recessive
ADP ANT1 dGK dGK ATP NDPK NDPK dG dG dGMP dGMP dGDP dGDP dGTP dGTP POLG POLG dA dA dAMP dAMP dADP dADP dATP dATP mtDNA mtDNA NDPK NDPK TK2 TK2 dC dC dGMP dCMP dGDP dCDP dCTP dGTP POLG POLG thymidine thymidine dAMP dTMP dTDP dADP dTTP dATP mitochondrion mitochondrion TK1 TK1 dTMP dTMP nDNA nDNA thymidine thymidine Thymidylate Thymidylate TP TP synthase synthase thymine thymine dUMP dUMP cytoplasm cytoplasm DNC MPV17
Mechanism leading to mtDNA mutations • Nucleotide imbalance cause mis-incorporation • Lack of DNA repair • Acceleration of DNA polymerase g activity by increased conc of dTTP Nishigaki Y et al. J Clin Invest. 2003;111:1913-1921
Why Mitochondrial DNA ? • Mito dNTP pools are physically separate and are regulated independently • More vulnerable to toxic effects of excessive dT because mtDNA is more dependent on dT SALVAGE pathway • Lack of an efficient mismatch repair system Nishigaki Y et al. J Clin Invest. 2003;111:1913-1921
Cytochrome c Oxidase, (Complex IV) Assembly requires a series of factors: SURF1 SCO2 SCO1 COX10 COX15 LRPPRC