Download
1 / 29
300 likes | 547 Views
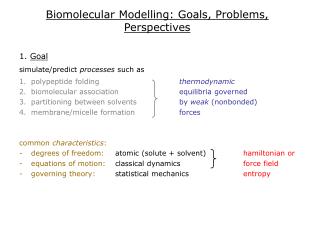
Biomolecular Modelling: Goals, Problems, Perspectives. 1. Goal simulate/predict processes such as polypeptide folding thermodynamic biomolecular association equilibria governed partitioning between solvents by weak (nonbonded) membrane/micelle formation forces

E N D
Biomolecular Modelling: Goals, Problems, Perspectives 1. Goal simulate/predict processes such as • polypeptide folding thermodynamic • biomolecular association equilibria governed • partitioning between solvents by weak (nonbonded) • membrane/micelle formation forces common characteristics: • degrees of freedom: atomic (solute + solvent) hamiltonian or • equations of motion: classical dynamics force field • governing theory: statistical mechanics entropy
Processes: Thermodynamic Equilibria Folding Micelle Formation folded/native denatured micelle mixture Complexation Partitioning in membrane bound unbound in water in mixtures
Methods to Compute Free Energy • Classical Statistical Mechanics: • Free Energy: • Free Energy Differences: • between two systems: and • depending on a parameter: • along a (phase space) coordinate:
Methods to Compute Free Energy • Counting of Configurations: • one simulation, but sufficient events sampled? • Thermodynamic Integration • many simulations: ensemble average <…> for each l value • then numerical integration • Perturbation Formula • one simulation, sufficient overlap?
DF stateA stateB Free Energy Difference via Thermodynamic Integration • Accurate: sufficient sampling <…> • sufficient l-points li • many (10 – 100) separate • simulations • for each new pair of states A and B a • new set of simlulations is required • for each • the state is unphysical • Very time consuming
Free Energy Calculations One-step perturbation technique and efficient sampling of relevant configurations Thermodynamic Integration N inhibitors: unbound bound 2 M Nsimulations 1010 200
Free Energy Calculations One-step Perturbation 2 simulations of an unphysical state which is chosen to optimise sampling for entire set ofN inhibitors Idea: use soft-core atoms for each site where the inhibitors possess different (or no) atoms The reference state simulation (R) should produce an ensemble that contains low-energy configurations for all of the Hamiltonians (inhibitors) H1, H2, … ,HN
A B’ B l0 l1 l2 l3 l4 l5 l6 l7 l8 A B A R B D C E conformational space
H2O Protein DGAbind A A DGARH2O Unphysical Reference Ligand R Unphysical Reference Ligand R DGBRH2O DGBbind B B C C … … DDGAB = DGBbind – DGAbind = DGARH2O – DGARprotein – DGBRH2O + DGBRprotein
Y1 (C) Y2 (T) Y3 Y4 Y5 Y6 Y7 Y8 Y9 Y10 U1 (A) U4 U5 U6 U7 U2 (G) U3 U8 U9 U10 U11 U12 U13 Free energies of base insertion, stacking, pairing in DNA
Five MD simulations to obtain free energies of base insertion, stacking, pairing 2.0 ns 3.4 ns 2.0 ns 2.0 ns (CGCGAXYTCGCG)
Free energy of insertion and stacking for particular pairs of central bases
A B C Stacking of adjacent central bases A, G, C, T A, U13, C, T A, G, Y9, T
Free energies of double base pairing in (CGCGAXYTCGCG)2 • 10x13x10x13 – 1 = 15899 values (in fact we did 1024) • U1-2 – Y1-2 and U1-13 – Y1-10, and vice versa (520 free energies) • Decompose the double free energies of pairing into single free energies of pairing purine pyrimidine
U10 Y7 U2 (G) Y9 14 kJ/mol 14 kJ/mol Y4 U4 Y4 U2 (G) 65 kJ/mol 99 kJ/mol Y10 U5 105 kJ/mol
Five MD simulations to obtain free energies of base insertion, stacking, pairing 2.0 ns 3.4 ns 2.0 ns 2.0 ns (CGCGAXYTCGCG)
Free Enthalpy of Solvation by Thermodynamic Integration Make Hamiltonian (Interaction) dependent on a coupling parameter l solute-solute assume = 0 (for simplicity) solute-solvent small solvent-solvent very large l=0 no solute-solvent interaction (solute in gas phase) l=1 full solute-solvent interaction (solute in solution)
(Free) Enthalpy and Entropy of Solvation difficult to calculate due to Uvv same term assumed: only solute-solvent interaction Uuv(l) depends on l solvent-solvent term Uvv does not
(Free) Enthalpy and Entropy of Solvation Uvv terms are absent g computable Calculate instead of DHS and TDSS: both computable yield insight into enthalpic and entropic effects
(Free) Enthalpy and Entropy of Solvation mole fraction Nico van der Vegt reference: J. Phys. Chem. B. (2004)
Solvation of Methane in Na+Cl- Solutions free enthalpy Na+Cl- methane solvation in salt DDU*uv triangles TDDS*uv squares relative to neat water energy (enthalpy) entropy concentration Entropy disfavours solvation increasingly with salt concentration (non-linear).
Solvation of Methane in Acetone Solution methane solvation in acetone DDU*uv triangles TDDS*uv squares relative to neat water: SPC water SPC/E water free enthalpy entropy energy (enthalpy) Entropy favours solvation. mole fraction
Solvation of Methane in Dimethylsulfoxide (DMSO) Solutions entropy energy (enthalpy) free enthalpy mole fraction mole fraction Energy favours solvation (non-linearly). reference: J. Chem. Phys. B. (2004)
DGSDUuvTDSuv Relative to Solvation in Pure Water mole fraction i different models dominant counteracts enthalpy enthalpy and entropy co-act counteract changes sign enthalpy relative and absolute contributions do vary entropy relative values of DDUuv, TDDSuvchange, DDGs not so much
Computer-aided Chemistry: ETH Zuerich Molecular Simulation Package GROMOS = Groningen Molecular Simulation + GROMOS Force Field Generally available:http://www.igc.ethz.ch/gromos Research Topics • searching conformational space • force field development • atomic • polarization • long range Coulomb • techniques to compute free energy • 3D structure determination • NMR data • X-ray data • quantum MD: reactions • solvent mixtures, partitioning • interpretation exp. data • applications • proteins, sugar, DNA, RNA, lipids, membranes, polymers • protein folding, stability • ligand binding • enzyme reactions
Computer-aided Chemistry: ETH Zuerich Group members Dirk Bakowies Indira Chandrasekhar David Kony Merijn Schenk (Alex de Vries) (Thereza Soares) (Nico van der Vegt) (Christine Peter) Alice Glaettli Yu Haibo Chris Oostenbrink Peter Gee Markus Christen Riccardo Baron Daniel Trzesniak Daan Geerke Bojan Zagrovic





















