3-Dimensional Crystal Structure
3-Dimensional Crystal Structure. 3-Dimensional Crystal Structure. 3-D Crystal Structure BW, Ch. 1; YC, Ch. 2; S, Ch. 2. General : A crystal structure is DEFINED by primitive lattice vectors a 1 , a 2 , a 3 .


3-Dimensional Crystal Structure
E N D
Presentation Transcript
3-D Crystal StructureBW, Ch. 1; YC, Ch. 2; S, Ch. 2 • General: A crystal structure is DEFINED by primitive lattice vectorsa1, a2, a3. • a1, a2, a3depend on geometry. Once specified, the primitive lattice structureis specified. • The lattice is generated by translating through a DIRECT LATTICE VECTOR: r = n1a1+n2a2+n3a3. (n1,n2,n3) are integers. rgenerates the lattice points. Each lattice point corresponds to a set of (n1,n2,n3).
Basis(or basis set) The set of atoms which, when placed at each lattice point, generates the crystal structure. • Crystal Structure Primitive lattice structure + basis. Translate the basis through all possible lattice vectors r = n1a1+n2a2+n3a3to get the crystal structure of the DIRECT LATTICE
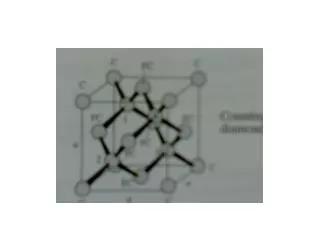
Diamond & Zincblende Structures • We’ve seen: Many common semiconductors have Diamond or Zincblende crystal structures Tetrahedral coordination:Each atom has 4 nearest-neighbors (nn). Basis set: 2 atoms. Primitive lattice face centered cubic (fcc). Diamond or Zincblende 2 atoms per fcc lattice point. Diamond: The 2 atoms are the same. Zincblende: The 2 atoms are different. The Cubic Unit Cell looks like
Zincblende/Diamond Lattices Diamond Lattice The Cubic Unit Cell Zincblende Lattice The Cubic Unit Cell Other views of the cubic unit cell
Diamond Lattice Diamond Lattice The Cubic Unit Cell
Zincblende (ZnS) Lattice Zincblende Lattice The Cubic Unit Cell.
View of tetrahedral coordination & 2 atom basis: Zincblende/Diamond face centered cubic (fcc) lattice with a 2 atom basis
Wurtzite Structure • We’ve also seen: Many semiconductors have the Wurtzite Structure Tetrahedral coordination: Each atom has 4 nearest-neighbors (nn). Basis set: 2 atoms. Primitive lattice hexagonal close packed (hcp). 2 atoms per hcp lattice point A Unit Cell looks like
Wurtzite Lattice View of tetrahedral coordination & 2 atom basis. Wurtzite hexagonal close packed (hcp) lattice, 2 atom basis
Diamond & Zincblende crystals • The primitive lattice is fcc. The fcc primitive lattice is generated byr = n1a1+n2a2+n3a3. • The fcc primitive lattice vectors are: a1 = (½)a(0,1,0), a2 = (½)a(1,0,1), a3 = (½)a(1,1,0) NOTE:Theai’s are NOTmutually orthogonal! Diamond: 2 identical atoms per fcc point Zincblende: 2 different atoms per fcc point Primitive fcc lattice cubic unit cell
primitive lattice points Wurtzite Crystals • The primitive lattice is hcp. The hcp primitive lattice is generated by r = n1a1 + n2a2 + n3a3. • The hcp primitive lattice vectors are: a1 = c(0,0,1) a2 = (½)a[(1,0,0) + (3)½(0,1,0)] a3 = (½)a[(-1,0,0)+ (3)½(0,1,0)] NOTE! Theseare NOTmutually orthogonal! • Wurtzite Crystals 2 atoms per hcp point Primitive hcp lattice hexagonal unit cell
Reciprocal LatticeReview? BW, Ch. 2; YC, Ch. 2; S, Ch. 2 • Motivations: (More discussion later). • The Schrödinger Equation & wavefunctions ψk(r). The solutions for electrons in a periodic potential. • In a 3d periodic crystal lattice, the electron potential has the form: V(r) V(r + R)R is the lattice periodicity • It can be shown that, for this V(r), wavefunctions have the form: ψk(r)= eikr uk(r), where uk(r) = uk(r+R). ψk(r) Bloch Functions • It can also be shown that, for r points on the direct lattice, the wavevectors k points on a lattice also Reciprocal Lattice
Reciprocal Lattice:A set of lattice points defined in terms of the (reciprocal) primitive lattice vectors b1, b2, b3. • b1, b2, b3are definedin terms of the direct primitive lattice vectors a1, a2, a3as bi 2π(aj ak)/Ω i,j,k, = 1,2,3 in cyclic permutations, Ω = direct lattice primitive cell volume Ω a1(a2 a3) • The reciprocal lattice geometry clearly depends on direct lattice geometry! • The reciprocal lattice is generated by forming all possible reciprocal lattice vectors:(ℓ1, ℓ2, ℓ3 = integers) K = ℓ1b1+ ℓ2b2 + ℓ3b3
The First Brillouin Zone (BZ) The region in k space which is the smallest polyhedron confined by planes bisecting the bi’s • The symmetry of the 1st BZ is determined by the symmetry of direct lattice. It can easily be shown that: The reciprocal lattice to the fcc direct lattice is the body centered cubic (bcc) lattice. • It can also be easily shown that the bi’s for this are b1 = 2π(-1,1,1)/a b2 = 2π(1,-1,1)/a b3 = 2π(1,1,1)/a
The 1st BZ for the fcc lattice(the primitive cell for the bcc k space lattice) looks like: b1 = 2π(-1,1,1)/a b2 = 2π(1,-1,1)/a b3 = 2π(1,1,1)/a
For the energy bands: Now discuss the labeling conventions for the high symmetry BZ points Labeling conventions The high symmetry pointson the BZ surfaceRoman letters The high symmetrydirections inside the BZ Greek letters The BZ CenterΓ(0,0,0) The symmetry directions: [100] ΓΔX , [111] ΓΛL , [110] ΓΣK We need to know something about these to understand how to interpret energy bandstructure diagrams:Ek vs k
Detailed View of BZ for Zincblende Lattice [110] ΓΣK [100] ΓΔX [111] ΓΛL To understand & interpret bandstructures, you need to be familiar with the high symmetry directions in this BZ!
The fcc 1st BZ: Has High Symmetry!A result of the high symmetry of direct lattice • The consequences for the bandstructures: If 2 wavevectors k & k in the BZ can be transformed into each other by a symmetry operation They are equivalent! e.g. In the BZ figure: There are 8 equivalent BZ faces When computing Ek one need only compute it for one of the equivalent k’s Using symmetry can save computational effort.
Consequences of BZ symmetries for bandstructures: Wavefunctions ψk(r) can be expressed such that they have definite transformation properties under crystal symmetry operations. QM Matrix elements of some operators O: such as <ψk(r)|O|ψk(r)>, used in calculating probabilities for transitions from one band to another when discussing optical & other properties (later in the course), can be shown by symmetry to vanish: So, some transitions are forbidden. This gives OPTICAL & other SELECTION RULES
Math of High Symmetry • The Math tool for all of this is GROUP THEORY This is an extremely powerful, important tool for understanding & simplifying the properties of crystals of high symmetry. • 22 pages in YC (Sect. 2.3)! • Read on your own! • Most is not needed for this course! • However, we will now briefly introduce some simple group theory notation & discuss some simple, relevant symmetries.
Group TheoryNotation: Crystal symmetry operations (which transform the crystal into itself) Operations relevant for the diamond & zincblende lattices: E Identity operation Cn n-fold rotationRotationby (2π/n) radians C2 = π (180°), C3 = (⅔)π (120°), C4 = (½)π (90°), C6= (⅓)π (60°) σReflectionsymmetry through a plane i Inversion symmetry Sn Cnrotation, followed by a reflection through a plane to therotation axis σ, I, Sn “Improper rotations” Also: All of these have inverses.
Crystal Symmetry Operations • For Rotations:Cn,we need to specify the rotation axis. • For Reflections:σ, we need to specify reflection plane • We usually use Miller indices (from SS physics) k, ℓ, n integers For Planes:(k,ℓ,n) or (kℓn): The plane containing the origin & is to the vector [k,ℓ,n] or [kℓn] For Vector directions:[k,ℓ,n] or [kn]: The vector to the plane (k,ℓ,n) or (kℓn) Also: k (bar on top)- k, ℓ (bar on top)-ℓ, etc.
Rotational Symmetries of the CH4 MoleculeThe Td Point Group. The same as for diamond & zincblende crystals
Diamond & Zincblende Symmetries ~ CH4 • HOWEVER, diamond has even more symmetry, since the 2 atom basis is made from 2 identical atoms. The diamond lattice has more translational symmetry than the zincblende lattice
Group Theory • Applications: It is used to simplify the computational effort necessary in the highly computational electronic bandstructure calculations.