MDD to MDR Transition
20 likes | 35 Views
Why is the modification in the regulation (MDD to MDR Transition) adapted?<br>History: You may be aware of the breast implant scandal in France wherein instead of medical grade Silicon, a low-quality Silicon was used as a raw material for breast implants which got ruptured and there were several complaints filed which led to the recall of the device by the manufacturer.<br>Read more@ https://iziel.com/mdd-to-mdr/

MDD to MDR Transition
E N D
Presentation Transcript
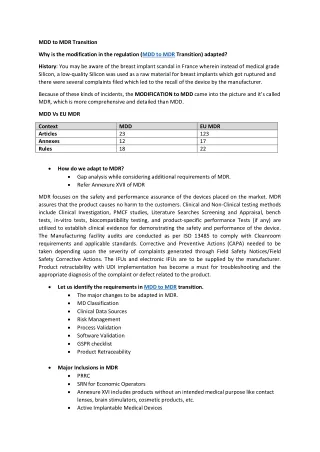
MDD to MDR Transition Why is the modification in the regulation (MDD to MDR Transition) adapted? History: You may be aware of the breast implant scandal in France wherein instead of medical grade Silicon, a low-quality Silicon was used as a raw material for breast implants which got ruptured and there were several complaints filed which led to the recall of the device by the manufacturer. Because of these kinds of incidents, the MODIFICATION to MDD came into the picture and it’s called MDR, which is more comprehensive and detailed than MDD. MDD Vs EU MDR Context Articles Annexes Rules MDD 23 12 18 EU MDR 123 17 22 How do we adapt to MDR? Gap analysis while considering additional requirements of MDR. Refer Annexure XVII of MDR MDR focuses on the safety and performance assurance of the devices placed on the market. MDR assures that the product causes no harm to the customers. Clinical and Non-Clinical testing methods include Clinical Investigation, PMCF studies, Literature Searches Screening and Appraisal, bench tests, in-vitro tests, biocompatibility testing, and product-specific performance Tests (if any) are utilized to establish clinical evidence for demonstrating the safety and performance of the device. The Manufacturing facility audits are conducted as per ISO 13485 to comply with Cleanroom requirements and applicable standards. Corrective and Preventive Actions (CAPA) needed to be taken depending upon the severity of complaints generated through Field Safety Notices/Field Safety Corrective Actions. The IFUs and electronic IFUs are to be supplied by the manufacturer. Product retractability with UDI implementation has become a must for troubleshooting and the appropriate diagnosis of the complaint or defect related to the product. Let us identify the requirements in MDD to MDR transition. The major changes to be adapted in MDR. MD Classification Clinical Data Sources Risk Management Process Validation Software Validation GSPR checklist Product Retraceability Major Inclusions in MDR PRRC SRN for Economic Operators Annexure XVI includes products without an intended medical purpose like contact lenses, brain stimulators, cosmetic products, etc. Active Implantable Medical Devices
Sterilization EN ISO 13485 and MDSAP Requirements Conformity Assessment Routes EUDAMED, UDI and Labelling Technical Documentation Clinical Evaluation Clinical Investigation and PMCF Reporting of serious incidents or failures to member states and trend reporting IFU and eIFU Requirements Cybersecurity SSCP linked to EUDAMED open to all end-users (including a layman) GSPR We at IZiel Healthcare have a long-standing collaboration withObelis,a Belgium-based (European Authorized Representatives) to provide a “One-Stop Solution” to fully support Class I, IIa, IIb & III medical device manufacturers across USA, Europe & Asia. This collaboration would ensure to obtain conformity with the MDR (2017/745) requirements and maintain the CE Marking of global medical devices through technical support, consultancy, representation, and device registration services.