Download

1 / 80
920 likes | 1.86k Views
PCR . Mr. S.Ghosh MITS Division of Biotechnology. Laboratory Tools and Techniques . The methods used by molecular biologists to study DNA have been developed through adaptation of the chemical reactions and biological processes that occur naturally in cells

E N D
PCR Mr. S.Ghosh MITS Division of Biotechnology
Laboratory Tools and Techniques The methods used by molecular biologists to study DNA have been developed through adaptation of the chemical reactions and biological processes that occur naturally in cells Many of the enzymes that copy DNA, make RNA from DNA, and synthesize proteins from an RNA template were first characterized in bacteria. This basic research has become fundamental to our understanding of the function of cells and have led to immense practical applications for studying a gene and its corresponding protein. As science advances, so do the number of tools available that are applicable to the study of molecular genetics.
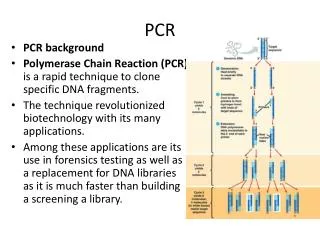
PCR The Polymerase Chain Reaction (PCR) provides an extremely sensitive means of amplifying relatively large quantities of DNA First described in 1985, Nobel Prize for Kary Mullis in 1993 The technique was made possible by the discovery of Taq polymerase, the DNA polymerase that is used by the bacterium Thermus aquaticus that was discovered in hot springs The primary materials, or reagents, used in PCR are: - DNA nucleotides, the building blocks for the new DNA - Template DNA, the DNA sequence that you want to amplify - Primers, single-stranded DNAs between 20 and 50 nucleotides long (oligonucleotides) that are complementary to a short region on either side of the template DNA - DNA polymerase, a heat stable enzyme that drives, or catalyzes, the synthesis of new DNA
PCR The cycling reactions : There are three major steps in a PCR, which are repeated for 20 to 40 cycles. This is done on an automated Thermo Cycler, which can heat and cool the reaction tubes in a very short time. Denaturation at around 94°C : During the denaturation, the double strand melts open to single stranded DNA, all enzymatic reactions stop (for example the extension from a previous cycle). Annealing at around 54°C : Hydrogen bonds are constantly formed and broken between the single stranded primer and the single stranded template. If the primers exactly fit the template, the hydrogen bonds are so strong that the primer stays attached Extension at around 72°C : The bases (complementary to the template) are coupled to the primer on the 3' side (the polymerase adds dNTP's from 5' to 3', reading the template from 3' to 5' side, bases are added complementary to the template)
PCR The different steps of PCR
PCR Exponential increase of the number of copies during PCR
PCR Every cycle results in a doubling of the number of strands DNA present After the first few cycles, most of the product DNA strands made are the same length as the distance between the primers The result is a dramatic amplification of a the DNA that exists between the primers. The amount of amplification is 2 raised to the n power; n represents the number of cycles that are performed. After 20 cycles, this would give approximately 1 million fold amplification. After 40 cycles the amplification would be 1 x 1012
PCR and Contamination The most important consideration in PCR is contamination Even the smallest contamination with DNA could affect amplification For example, if a technician in a crime lab set up a test reaction (with blood from the crime scene) after setting up a positive control reaction (with blood from the suspect) cross contamination between the samples could result in an erroneous incrimination, even if the technician changed pipette tips between samples. A few blood cells could volitilize in the pipette, stick to the plastic of the pipette, and then get ejected into the test sample Modern labs take account of this fact and devote tremendous effort to avoiding cross-contamination
Optimizing PCR protocols PCR can be very tricky While PCR is a very powerful technique, often enough it is not possible to achieve optimum results without optimizing the protocol Critical PCR parameters: - Concentration of DNA template, nucleotides, divalent cations (especially Mg2+) and polymerase - Error rate of the polymerase (Taq, Vent exo, Pfu) - Primer design
Perhaps the most critical parameter for successful PCR is the design of primers Primer length - specificity and the temperature of annealing are at least partly dependent on primer length - oligonucleotides between 20 and 30 (50) bases are highly sequence specific - primer length is proportional to annealing efficiency: in general, the longer the primer, the more inefficient the annealing - the primers should not be too short as specificity decreases Primer design General notes on primer design in PCR Primer selection Critical variables are: - primer length - melting temperature (Tm) - specificity - complementary primer sequences - G/C content - 3’-end sequence
Primer design Specificity Primer specificity is at least partly dependent on primer length: there are many more unique 24 base oligos than there are 15 base pair oligos Probability that a sequence of length n will occur randomly in a sequence of length m is: Example: the mtDNA genome has about 20,000 bases, the probability of randomly finding sequences of length n is: n Pn 5 19.52 10 1.91 x 10-2 15 1.86 x 10-5 P = (m – n +1) x (¼)n
Primer design • Complementary primer sequences • primers need to be designed with absolutely no intra-primer homology beyond 3 base pairs. If a primer has such a region of self-homology, “snap back” can occur • - another related danger is inter-primer homology: partial homology in the middle regions of two primers can interfere with hybridization. If the homology should occur at the 3' end of either primer, primer dimer formation will occur • G/C content • ideally a primer should have a near random mix of nucleotides, a 50% GC content • there should be no PolyG or PolyC stretches that can promote non-specific annealing 3’-end sequence - the 3' terminal position in PCR primers is essential for the control of mis-priming - inclusion of a G or C residue at the 3' end of primers helps to ensure correct binding (stronger hydrogen bonding of G/C residues)
Primer design Melting temperature (Tm) - the goal should be to design a primer with an annealing temperature of at least 50°C - the relationship between annealing temperature and melting temperature is one of the “Black Boxes” of PCR - a general rule-of-thumb is to use an annealing temperature that is 5°C lower than the melting temperature - the melting temperatures of oligos are most accurately calculated using nearest neighbor thermodynamic calculations with the formula: Tm = H [S+ R ln (c/4)] –273.15 °C + 16.6 log 10 [K+] (H is the enthalpy, S is the entropy for helix formation, R is the molar gas constant and c is the concentration of primer) - a good working approximation of this value can be calculated using the Wallace formula: Tm = 4x (#C+#G) + 2x (#A+#T) °C - both of the primers should be designed such that they have similar melting temperatures. If primers are mismatched in terms of Tm, amplification will be less efficient or may not work: the primer with the higher Tm will mis-prime at lower temperatures; the primer with the lower Tm may not work at higher temperatures.
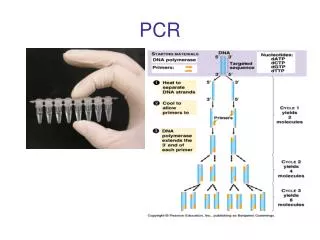
Typical components of a PCR include: • DNA: the template used to synthesize new DNA strands. • DNA polymerase: an enzyme that synthesizes new DNA strands. • Two PCR primers: short DNA molecules (oligonucleotides) that define the DNA sequence to be amplified. • Deoxynucleotidetriphosphates (dNTPs): the building blocks for the newly synthesized DNA strands. • Reaction buffer: a chemical solution that provides the optimal environmental conditions. • Magnesium: a necessary cofactor for DNA polymerase activity. The PCR Process—Reaction Components
One PCR cycle consists of a DNA denaturation step, a primer annealing step and a primer extension step. DNA Denaturation: Expose the DNA template to high temperatures to separate the two DNA strands and allow access by DNA polymerase and PCR primers. Primer Annealing: Lower the temperature to allow primers to anneal to their complementary sequence. Primer E xtension: Adjust the temperature for optimal thermostable DNA polymerase activity to extend primers. • PCR uses a thermostable DNA polymerase so that the DNA polymerase is not heat-inactivated during the DNA denaturation step. Taq DNA polymerase is the most commonly used DNA polymerase for PCR. HPCR Amplify DNA?
DNA polymerase extends the primer by sequentially adding a single dNTP (dATP, dGTP, dCTP or dTTP) that is complementary to the existing DNA strand • The sequence of the newly synthesized strand is complementary to that of the template strand. • The dNTP is added to the 3´ end of the growing DNA strand, so DNA synthesis occurs in the 5´ to 3´ direction. Mechanism of DNA Synthesis
Thermal cyclers have a heat-conducting block to modulate reaction temperature. • Thermal cyclers are programmed to maintain the appropriate temperature for the required length of time for each step of the PCR cycle. • Reaction tubes are placed inside the thermal cycler, which heats and cools the heat block to achieve the necessary temperature. Instrumentation
A typical thermal cycler program is: Initial DNA denaturation at 95°C for 2 minutes 20–35 PCR cycles: Denaturation at 95°C for 30 seconds to 1 minute Annealing at 42–65°C for 1 minute Extension at 68–74°C for 1–2 minutes Final extension at 68–74°C for 5–10 minutes Soak at 4°C Thermal Cycling Programs
Many PCR parameters might need to be optimized to increase yield, sensitivity of detection or amplification specificity. These parameters include: • Magnesium concentration • Primer annealing temperature • PCR primer design • DNA quality • DNA quantity PCR Optimization
Magnesium concentration is often one of the most important factors to optimize when performing PCR. • The optimal Mg2+ concentration will depend upon the primers, template, DNA polymerase, dNTP concentration and other factors. • Some reactions amplify equally well at a number of Mg2+ concentrations, but some reactions only amplify well at a very specific Mg2+ concentration. • When using a set of PCR primers for the first time, titrate magnesium in 0.5 or 1.0mM increments to empirically determine the optimal Mg2+ concentration. Magnesium Concentration
PCR primers must anneal to the DNA template at the chosen annealing temperature. • The optimal annealing temperature depends on the length and nucleotide composition of the PCR primers • The optimal annealing temperature is often within 5°C of the melting temperature (Tm) of the PCR primer The melting temperature is defined as the temperature at which 50% of complementary DNA molecules will be annealed (i.e., double-stranded). • When performing multiplex PCR, where multiple DNA targets are amplified in a single PCR, all sets of PCR primers must have similar annealing temperatures. Primer Annealing Temperature
DNA should be intact and free of contaminants that inhibit amplification. • Contaminants can be purified from the original DNA source. • Heme from blood, humic acid from soil and melanin from hair • Contaminants can be introduced during the purification process. • Phenol, ethanol, sodium dodecyl sulfate (SDS) and other detergents, and salts. DNA Quality
DNA quantity • More template is not necessarily better. • Too much template can cause nonspecific amplification. • Too little template will result in little or no PCR product. • The optimal amount of template will depend on the size of the DNA molecule. DNA Quantity
PCR and RT-PCR have hundreds of applications. In addition to targeting and amplifying a specific DNA or RNA sequence, some common uses include: • Labeling DNA or RNA molecules with tags, such as fluorophores or radioactive labels, for use as tools in other experiments. • Cloning a DNA or RNA sequence • Detecting DNA and RNA • Quantifying DNA and RNA • Genotyping and DNA-based identification Applications of PCR
Labeling DNA with tags for use as tools (probes) to visualize complementary DNA or RNA molecules. • Radioactive labels. • Radioactively labeled probes will darken an X-ray film. • Fluorescent labels (nonradioactive) • Fluors will absorb light energy of a specific wavelength (the excitation wavelength) and emit light at a different wavelength (emission wavelength). • The emitted light is detected by specialized instruments such as fluorometers. Labeling DNA
Avoids problems associated with the plateau effect, which reduces amplification efficiency and limits the amount of PCR product generated due to depletion of reactants, inactivation of DNA polymerase and accumulation of reaction products. • The result of the plateau effect is that the amount of PCR product generated is no longer proportional to the amount of DNA starting material. • The plateau effect becomes more pronounced at higher cycle numbers. • Often performed in real time to monitor the accumulation of PCR product at each cycle. • Real-time PCR allows scientists to quantify DNA before the plateau effect begins to limit PCR product synthesis. Quantitative PCR
THE PROBLEM • NEED TO QUANTITATE DIFFERENCES IN mRNA EXPRESSION • SMALL AMOUNTS OF mRNA • LASER CAPTURE • SMALL AMOUNTS OF TISSUE • PRIMARY CELLS • PRECIOUS REAGENTS
THE PROBLEM • QUANTITATION OF mRNA • northern blotting • ribonuclease protection assay • in situ hybridization • PCR • most sensitive • can discriminate closely related mRNAs • technically simple • but difficult to get truly quantitative results using conventional PCR
Ratio target gene in experimental/control = fold change in target gene fold change in reference gene NORTHERN control expt 10X target gene internal control gene actin, GAPDH, RPLP0 etc 2X Corrected fold increase = 10/2 = 5
same copy number in all cells • expressed in all cells • medium copy number advantageous • correction more accurate Standards
REAL TIME PCR • kinetic approach • early stages • while still linear www.biorad.com
3. intensifier 5. ccd detector 350,000 pixels 1. halogen tungsten lamp 2b. emission filters 2a. excitation filters 4. sample plate
threshold Ct SERIES OF 10-FOLD DILUTIONS
PFAFFL METHOD • M.W. Pfaffl, Nucleic Acids Research 2001 29:2002-2007
AFTER 1 CYCLE 100% = 2.00x 90% = 1.90x 80% = 1.80x 70% = 1.70x
AFTER 1 CYCLE 100% = 2.00x 90% = 1.90x 80% = 1.80x 70% = 1.70x AFTER N CYCLES: fold increase = (efficiency)n