Download
1 / 21
700 likes | 2.51k Views
5. Vibrations in Molecules. Introduction 5.1. The vibrations of diatomic molecules 5.1.1 Molecular vibrations 5.1.2 Selection rules in IR spectroscopy 5.1.3 Anharmonicity 5.2. The vibrations of polyatomic molecules 5.2.1 Normal modes 5.2.2 Vibrational IR spectra.

E N D
5. Vibrations in Molecules Introduction 5.1. The vibrations of diatomic molecules 5.1.1 Molecular vibrations 5.1.2 Selection rules in IR spectroscopy 5.1.3 Anharmonicity 5.2. The vibrations of polyatomic molecules 5.2.1 Normal modes 5.2.2 Vibrational IR spectra
Electronic transitions Vibrational transitions Rotational transitions
Vibrations of molecules can be studied via Infra-Red or Raman spectroscopies. Heated ceramic filament: Black body All frequencies Only one frequency The arrangement adopted in Raman spectroscopy. Here, this is the scattered radiation that is observed at 90º from the incident beam. The molecules scatter the incident radiation and give or absorb a part of their vibrational energies. The scattered radiation can then have lower (absorb) and higher (give) energy than the incident radiation. The layout of a typical Infra-red absorption spectrometer. From the comparison between the reference beam and the one passing through the sample, we can deduce the frequencies absorbed by the excitation of molecules in their vibrational energy levels.
5.1. The vibrations of diatomic molecules 5.1.1 Molecular vibrations A. The harmonic approximation: x= R-Re = the deformation of the bond k= the force constant of the bond Zero of energy: V(0)=0 Energy minimum
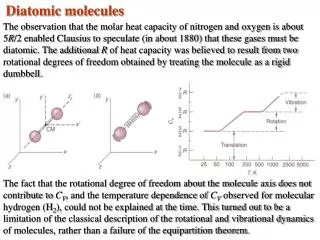
The steeper the walls of the potential (the stiffer the bond), the greater the force constant. B. The Schrödinger equation In Chap 3: the motion of a system composed of 2 bodies was dissociated in the motion for the center of mass and the relative motion of one body with respect to the other one. The relative motion of 2 atoms of masses m1 and m2 with a parabolic potential energy is described with this SE: meff is the effective mass (or reduced mass for a diatomic molecule) In chap 2, we have studied the motion of a particle in a harmonic potential. We can apply the same solution for the vibration of two bodies by considering that it is a particle of mass meff that is in a harmonic potential. The eigenvalues or permittedvibrational energy levelsare:
With an harmonic potential, the levels are equally spaced by ℏ with one another Vibrational terms of a molecule, i.e. the vibrational states G() expressed in wavenumber : E= hc G() Two limit cases for the effective mass: 1) Homonuclear diatomic molecule: m1= m2→meff = ½ m1 2) Heteronuclear diatomic molecule with m1 >> m2, like HI. mI >> mH→meff≃ mH → the I atom barely moves. The SE equation describes almost the motion an H atom attached to a wall.
Various notations are commonly used Wavenumber Pulsation Wavelength Planck constant
C. The form of the wavefunctions (see Chap 2) Hermite Polynomial Gaussian-type function N is the normalization constant The higher the quantum number , the larger the number of nodes in the wavefunction
+q -q Re 5.1.2 Selections rules A. Permanent Dipole moment An electric dipole consists of two electric charges q and -q separated by a distance R. This arrangement of charges is represented by a vector, the electric dipole moment with a magnitude: = Re q Unit: Debye, 1D = 3.33×10-30Cm When the molecule is at its equilibrium position, the dipole moment is called “permanent dipole moment” 0. All heteronuclear diatomic molecules are polar because their atoms do not have the same electronegativity . In a first approximation: /D≃ B. Selection rules The probability for a vibrational transition to occur, i.e. the intensity of the different lines in the IR spectrum, is given by the transition dipole momentfibetween an initial vibrational state i and a vibrational final state f :
+q -q R= Re+ x The electric dipole moment operator depends on the location of all electrons and nuclei, so its varies with the modification in the intermolecular distance “x”. The variation of with displacement of the nuclei from equilibrium is: 0 is the permanent dipole moment for the molecule in the equilibrium position Re. 0 The higher terms can be neglected for small displacements of the nuclei The two states i and f are orthogonal. Because they are solutions of the operator H which is Hermitian (see Chap 2: properties of hermitian operator)
First condition: fi= 0, if ∂/ ∂x = 0 Second condition: In order to have a vibrational transition visible in IR spectroscopy: the electric dipole moment of the molecule must change when the atoms are displacedrelative to one another. Such vibrations are “ infrared active”. It is valid for polyatomic molecules. By introducing the wavefunctions of the initial state i and final state f , which are the solutions of the SE for an harmonic oscillator (see chap 2), the following selection rulesis obtained: = ±1 Note 1: Vibrations in homonuclear diatomic molecules do not create a variation of not possible to study them with IR spectroscopy. Note 2: A molecule without a permanent dipole moment can be studied, because what is required is a variation of with the displacement. This variation can start from 0.
C. Classical explanation: If the oscillation frequency of the electric field of a radiation is similar to the frequency =/2 of one vibrational motion in a molecule (which involves a variation of the charge distribution), then the molecule can absorb the energy h of one photon from the radiation. Intuitively, we can see this absorption of energy like the resonance phenomenon in classical mechanics (Tacoma bridge). D. Consequences of the harmonic approximation At room T, molecules are mainly in their vibrational ground state. Hence, in IR absorption spectroscopy, the molecules are excited from the ground state to the first excited state: 0→1, since the selection rule is = ±1 the IR spectrum should contain only one line for diatomic molecule. At higher T, other transitions can occur: 2 →3 or 3 →4, but all of them need the absorption of a photon with the same energy, i.e. the absorption lines appear at the same frequency, because the energy between two states is constant. Although the main features are there, it is not exactly what shows the actual absorption spectra…. The potential is not harmonic. According to the harmonic oscillator, a chemical bond cannot break.
Tacoma Narrows Bridge Failure wind 8m 1974 The nature and severity of the torsional movement is revealed in this picture taken from the Tacoma end of the suspension span. When the twisting motion was at the maximum, elevation of the sidewalk at the right was 28 feet (8.5m) higher than the sidewalk at the left. This photograph shows the twisting motion of the center span just prior to failure. car A few minutes after the first piece of concrete fell, this 600 foot section broke out of the suspension span, turning upside down as it crashed in Puget Sound. Note how the floor assembly and the solid girders have been twisted and warped. The square object in mid air (near the centre of the photograph) is a 25 foot (7.6m) section of concrete pavement. Notice the car in the top right corner.
5.1.3 Anharmonicity An simple analytical expression, called the Morse potential, represents the main features of the real potential for a molecule: - close to the minimum potential of depth De, the potential is close to be harmonic. - for large displacement, the potential represents the bond dissociation The Schrödinger equation can be solved with the Morse potential and the permitted energy levels are: When increases, the second term becomes quickly more negative than the first term →the energy levels become less widely spaced at high excitation At high temperature, several weak lines appear. Moreover, the selection rule = ±1 indicates the more intense lines, but weak transitions of > 1 are now allowed.
5.2. The vibrations of polyatomic molecules 5.2.1 Normal modes A. Number of vibrational modes A molecule composed of N atoms. The total number of coordinates to specify the locations of N atoms is 3N (e.g: x, y, z per atom). But, we are interested in the relative motion of these atoms with respect to each other, →remove the 3 coordinates (Xc.m, Yc.m, Zc.m) of the center of gravity, which characterize the translation of the molecule. →remove the 3 angular coordinates (, , ) which specify the global rotation of the molecule in the space. (2 angles are enough for linear molecules) The remaining coordinates are directly related to the vibrations between atoms. There are 3N-6 displacements of the atoms relative to one another: these are the 3N-6 independent vibrationalmodes (3N-5 for linear molecules)
B. Combinations of displacements L Example: CO2 3 atoms, 3N-5= 4 modes 2 modes describe the variation of the CO bond lengths (stretching modes) and 2 modes the variations of the bond angles (bending modes). If the stretching modes are L and R (each bond is considered separately), then one see that if one is excited, it gives the energy to the other mode. The modes are not independent. It’s possible to find specific modes that are independent, that is if one is excited, it does not excite the other: these are the normal modes. For CO2, these are: 1, 2, 3 and 4. Each normal mode q behaves like an independent harmonic oscillator (approximation), so has a series of terms Gq() where is the wavenumber of the mode q and depends on the force constant kq and the effective mass of the mode mq. R Stretching symmetric 1 2 Stretching Anti-symmetric 3 4
Antisymmetric stretching Symmetric stretching 2 Bending modes of same frequency Evib(total)=E(v1)+E(v2)+E(v3)+E(v4) Example: Evib(total)=hv1+0+hv3+0 Simple addition of energy because the vibrational modes are independent
5.2.2 Vibrational IR spectra In general, a normal mode is a composite motion of simultaneous stretching and bending of bonds. The effective mass of a mode is a measure of the mass that is swung during the vibration. The effective mass is described from the masses of the atoms moving in this modes. The bending modes appears a lower frequency than the stretching modes. The wavenumber at which a transition appears is related to the force constant kq of the oscillator (curvature of the parabola), that is related to the stiffness of bonds involved in the vibration. IR and Raman spectroscopies can be used for chemical analysis, since chemical groups are composed of bonds with different stiffness.
E R Vibrational frequency: (C-C)[700-1200cm-1] < (C=C) [1620-1680cm-1]< (C≡C) [2100-2260cm-1] Bond lengths: Re(C-C)[1.54Å] > Re(C=C) [1.35Å]> Re(C ≡ C) [1.20Å] Bond dissociation energy: D0(C-C) [368 kJ/mol] < D0(C=C) [720 kJ/mol] < D0(C ≡ C) [962 kJ/mol] k(C-C) < k(C=C)< k(C ≡C)