Download

1 / 29
340 likes | 1.04k Views
Cystic Fibrosis. Paul Ricard , PT, DPT, CCS JoAnn Moriarty-Baron , PT, DPT UML Physical Therapy Department Cardiopulmonary PT Lab 34.614 April 28, 2011. Cystic Fibrosis.

E N D
Cystic Fibrosis Paul Ricard, PT, DPT, CCS JoAnn Moriarty-Baron, PT, DPT UML Physical Therapy Department Cardiopulmonary PT Lab 34.614 April 28, 2011
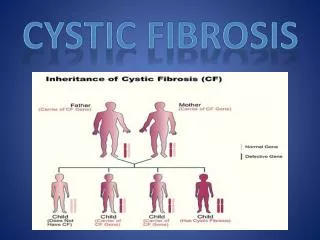
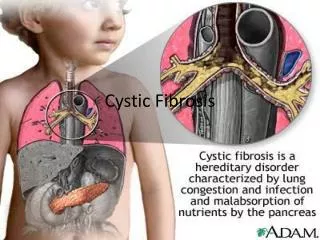
Cystic Fibrosis • Cystic Fibrosis is an inherited, autosomal recessive, multi-system health condition characterized by an abnormality in exocrine gland function.
Pathophysiology • There is an altered gene located on the long arm of chromosome 7 • A deletion of this protein leads to altered Cystic fibrosis transmembrane conductance regulator (CFTR)
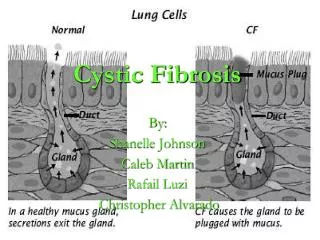
Pathophysiology • The defect effects chloride ion transmission across epithelial cells • Results in excessive sodium reabsorption and ↑ viscosity of the exocrine secretions • Leads to: • Dehydration of surface fluids • Abnormally salty sweat • Thick mucus that clogs tubes, tubules, ducts
Multisystem Involvement • Gastrointestinal • Vitamin deficiency • Malabsorption • Genitourinary • Blockages • Infertility • Integumentary • Dry skin / dehydration • Musculoskeletal • Muscle atrophy • Pulmonary • Cardiac • Endocrine • DM
Epidemiology • Common in populations of European origin • White Americans 1/1600 to 1/2000 live births • Estimated incidence range from 1/500 Amish (Ohio) to 1/90,000 Hawaiian Orientals • Equal gender distribution • Approximately 30,000 children and adults in the United States have cystic fibrosis • In US ~5% of the population carries a single CF gene
Most common lethal genetic disease of the Caucasian population ! Both parents must carry the genetic defect
The good news... Median survival rate is going up 1991 was ~20 years 1998 was ~ 32.3 years 2003 was ~32 years 2008 was ~ 37.4 years Currently >50% of children with CF survive to adulthood The bad news.... Expected decline in pulmonary function in patients with CF is between 2-3% per year 90% of those with CF die of respiratory failure
Diagnosis of CF • Commonly dx at birth, but may not be suspected until later in life • Most common diagnostic test for CF is the Sweat Test • Positive Sweat Test = • Chloride concentration >70mmol/liter • Sodium concentration >60 mmol/liter • (Normal values are 50 and 40 mmol/liter respectively) • Meconium ileus at birth has become hallmark sign for dx
Diagnosis of CF • Meconium ilieus • Constipation • Abdominal distension • Colicky, pain • Emesis • Rapid dehydration • Fe+ deficient Anemia
Other Signs used for Diagnosis • Familial hx • Exocrine pancreatic insufficiency • Chronic pulmonary changes • Problems with reproductive functions • Males are azoospermic • Females have ↓ fertilization
Characteristics of CF • Short stature with ↓body weight • Voracious appetites due to ↑ resting metabolic rate with intestinal malabsorption • Osteopenia / premature osteoporosis • Barrel chest with flattened diaphragm leads to a sense of fullness and ↓food intake
Pathological Process • CF may affect gas exchange in several ways: • Increase distance for gas exchange • clogged alveoli • chronically infected areas • Decrease size of gas exchange area • collapsed segments • shunted blood • Alter the partial pressure gradient • poor ventilation • poor distribution or air • elevation of arterial partial pressure
Pathological Process • Changes begin in the bronchioles • Decrease size of gas exchange area • Decreased surfactant secretion which decreases surface tension and increase alveolar collapse • Mucus plugs can also temporarily cause distal alveolar collapse
Pathological Process • As CF progresses larger, more central airways become involved • Usually infected with Staphylococcus aureus or Pseudomonas aeruginosa • Mucus secreting glands become hypertrophied • Increased secretions reduce lumen of airways • Normal mucociliary mechanism is impaired causing duct obstruction • Structure of airways are altered and bronchiectatic reconstruction occurs
Secondary impairments • Pulmonary hypertension • Right sided heart failure (cor pulmonale) • Pneumothorax • Hemoptysis
Medical Management • Treatment is driven at reducing sequelae of primary impairments to slow progression of health condition and increase quality of life • Pharmacotherapy • Antibiotics • Bronchodilators • Mucolytics • Anti-inflammatory medications • Gene therapy • Surgical procedures • Reduce dead space • Pleural sclerosis • Bronchial artery embolization • Lung transplant • 120 – 150 people/year • >90% alive after 1 yr, 50% alive after 5 yrs
PT Interventions • Preferred practice patterns: • 4A: Primary prevention/risk reduction for skeletal demineralization • 6C: impaired ventilation, respiratory/Gas exchange, and aerobic capacity/endurance associated with airway clearance dysfunction • 6E: impaired ventilation, respiratory/Gas exchange associated with ventilatory pump dysfunction or failure • 6F: impaired ventilation, respiratory/Gas exchange associated with respiratory failure
PT Interventions • Both aerobic and strength training increase pulmonary function, strength, and aerobic capacity, positively impact control of diabetes, body image, and levels of anxiety. • Exercise: • increases sputum clearance • delays onset of dyspnea • delays decline in pulmonary function • prevents decrease in bone density • enhances cellular immune response • increases feelings of well-being
Traditional PT Interventions Chest Physiotherapy (CPT) Postural Drainage (PD) Self-administered Techniques Active Cycles of Breathing (ACBT) Autogenic Drainage (AD) PT Interventions for Airway Clearance Techniques (ACT)
Active Cycle of Breathing Easy to learn Can be done anywhere Can be used with children Can be done in postural drainage positions Avoid cough until Huff during phase 3 Autogenic Drainage Requires concentration Person must be able to recognize location of secretions Less suitable for children Can be done anywhere Cough suppression until mucus is in larger airways Self Administered Techniques
Active Cycles of Breathing • Phase I – Breathing control • Diaphragmatic breathing / lower ribcage expansion (5-10 secs) • Phase II – Thoracic expansion • ↑ inspiration to max. volume, expiration is passive, relaxed • 3 sec inspiratory holds / sniff for collateral ventilation to reinflate atelectatic alveoli • Phase III – Forced expiration • Huffing alternated with breathing control • Medium volume huffs→breath control→high volume huff • Continue until huffs are non-productive for 2 consecutive cycles
Autogenic Drainage • Phase I – Unsticking • Deep inhale through nose exhales through mouth @ low lung volumes then breathes normally here until pt aware of secretions in small airways • Phase II – Collection • ↑ depth of inspiration to midlung volume with ↑ expiratory volume • Secretions passed to mid-size, central airways • Phase III –Evacuation • ↑ inspiration to high lung volumes →max inspiratory volume • High volume huff to expectorate mucus
Old (PD) vs. New (AD) clearance techniques • Both the AD and PD groups demonstrated improved pulmonary function with no significant difference between the two groups. • Patients with CF exhibited a marked preference for the AD technique. • Both AD and PD are effective methods of performing ACT for patients with CF • Benefits of either technique are enhanced by measures which encourage adherence.
Active Cycle Of Breathing Technique (ACBT) • Four studies, with four different comparators, found that ACBT was comparable to other therapies in outcomes such as patient preference, lung function, sputum weight, oxygen saturation, and number of pulmonary exacerbations.
Toys! • PEP • Oscillatory PEP • High frequency chest wall oscillation (Vest) • Intrapulmonary pressure ventilation (cough-a-lator)
Efficacy Of Airway Clearance Techniques • Systematic Review 1960-2004 to determine efficacy of different nonpharmacologic interventions for airway clearance. Postural drainage, chest wall percussion/vibration, and huffing (forced expiration techniques increased airway clearance as assessed by airway characteristics (volume, weight, viscosity) but long term effects were not clear. Devices and non-assisted techniques appeared to be just as effective at CPT in increasing sputum production.
Conclusions • No clear advantage of conventional CPT over other airway clearance techniques in terms of respiratory function • There was a trend for participants to prefer self-administered airway clearance techniques.
References • Deturk W, Cahalin L.Cardiovascular and Pulmonary Physical Therapy.An Evidence-based Approach. United States of America:McGraw-Hill Companies Inc;2004:465-467 • Orenstein DM, Higgins LW. Update on the role of exercise in cystic fibrosis. • Curr Opin Pulm Med 11:519-523. • 1. Bell SC, Morrie NR. Editorial: Exercise testing in patients with cystic fibrosis: • Why and which? Journal of Cystic Fibrosis 9 (2010);299-301. • 2. Larry’s book • Main E, Prasad A, van der Schans CP. Conventional chest physiotherapy • compared to other airway clearance techniques for cystic fibrosis (Review). • The Cochrrane Library 2009. Issue 2. • McCool FD, Rosen MJ. Nonpharmacological Airway Clearance Therapies: • ACCP Evidence-Based Clinical Practice Guidelines. CHEST 2006; • 129:250S–259.