Download

1 / 42
530 likes | 1.28k Views
CYSTIC FIBROSIS. "Woe is the child who tastes salty from a kiss on the brow, for he is cursed, and soon must die.”. -Northern European Folklore. CYSTIC FIBROSIS. Figure 8.

E N D
"Woe is the child who tastes salty from a kiss on the brow, for he is cursed, and soon must die.” -Northern European Folklore
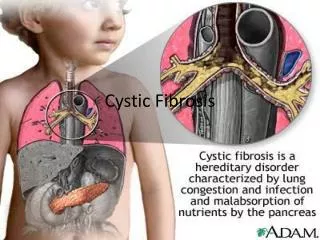
CYSTIC FIBROSIS Figure 8 Cystic Fibrosis (CF) is a lethal inherited disease that affects the lungs, digestive system, sweat glands, and male fertility Its name derives from the fibrous scar tissue which develops in the pancreas
CYSTIC FIBROSIS Cystic Fibrosis affects the body’s ability to move salt and water in and out of a cell This results in the lungs and the pancreas secreting a thick mucus This mucus blocks passageways and prevents proper function
Hallmarks of CF • Very salty-tasting skin • Appetite, but poor growth & weight gain • Coughing, wheezing & shortness of breath • Lung infections, e.g. pneumonia/bronchitis
Clinical Aspects Cystic fibrosis affects the entire body • Lungs and sinuses • GI, liver and pancreas • Endocrine system • Reproductive system
Epidemiology CF is a rare disease • Approximately 30,000 in the U.S. people have CF • Over 10 million Americans are unknowing carriers. • Around 2,500 children with CF are born each year. CF is a disease of Caucasians.
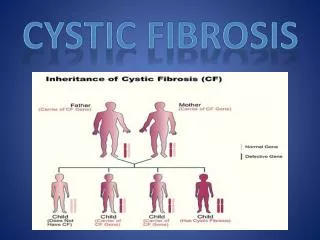
Heritability CF is a hereditary disease. • Unaffected parents can have children with CF. • Males and females are equally likely to be diagnosed.
Carrier Dad Carrier Mom Eggs Sperm Baby with CF Normal carrier Normal carrier Normal non-carrier
Signs and Symptoms • Thick, viscous mucus secretion in the lungs • Changes in color and amount of sputum • chronic coughing • Wheezing • Bronchitis • salty skin or poor growth • Weight loss (abdominal swelling)
Mapping the gene for CF • Gene linkage studies were able to map the mutation to chromosome 7. • Classical genetics techniques were not able to accurately pinpoint the mutated gene.
Gene Locus Chromosome 7; Locus 7q31.2 • The CFTR gene: • is 250, 000 bp long • contains 27 exons • the protein has 1, 480 amino acids with a molecular mass of 168, 138 Da • nucleotide of ~ 6,500
Mapping the gene for CF 1989: Lap-Chee Tsui, at the Hospital for Sick Children in Toronto, clones the CFTR gene. Victory tastes sweet. Chromosome walking and jumping techniques were used to identify and sequence the 180,000 bp gene.
The ΔF508 Mutation The mutation results in the deletion of a single amino acid (Phe) at position 508. A 3 base pair deletion called ΔF508 is the most common mutation causing cystic fibrosis
Benefits of ΔF508 The ΔF508 mutation most likely occurred over 50,000 years ago in Northern Europe. Individuals with two copies of ΔF508 get cystic fibrosis and often cannot reproduce. Having one copy of ΔF508reduces water loss during cholera, greatly increasing the chance of survival.
The Function of CFTR CFTR encodes a 170 kDa, membrane-based protein with an active transport function
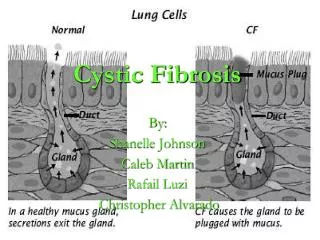
Role of CFTR Protein The CFTR protein plays a vital role in mucus function The CFTR protein helps to keep mucus from becoming thick
Role of CFTR Protein • In CF, the CFTR cannot allow chloride ions to move out of the mucus-producing cells • This means water doesn’t leave and it results in the mucus becoming thick • This in turn blocks the passageways, and allow bacteria to feed off the mucus, which results in more infections
From Mutation to Disease The mutant form of CFTR prevents chloride transport, causing mucus build-up Mucus clogs the airways and disrupts the function of the pancreas & intestines.
CFTR Mutations Over 1,000 mutations in CFTR have been found. ΔF508 accounts for just 70% of CF cases.
The Sweat Test • Measures the concentration of chloride and sodium that is excreted in sweat. • Two reliable positive results on two separate days is diagnostic for CF. • Clinical presentation, family history and patient age must be considered to interpret the results.
5 Classes of CFTR Mutations CF Mutations can be classified by the effect they have on the CFTR protein.
5 Classes of CFTR Mutations I Defective Production II Defective Processing III Defective Regulation IV Defective Conductance V Reduced Amounts
Genotype Class and Mortality Mutation class can affect disease mortality.
Genotype and Phenotype Clinical phenotypes can vary widely across mutations
Newborn Screening Infants can easily be diagnosed with a blood test • Elevated levels of trypsinogen indicate CF • Screening programs identify 10% of cases at birth Most hospitals do not screen for CF at birth. • Should they?
Genetic Carrier Testing • Tests for common CF mutations are available. • The type of defective CF gene can affect the type of CF symptoms. • However, genetic testing cannot fully determine how severe a person's CF will be in advance.
Carrier Screening • Population-based screening: • Particular genetic carrier tests offered to everyone in the general population • Targeted population-based screening: • Carrier screening limited to particular groups of people determined to be at higher risk for specific genetic disorders • e.g. Ethnicity-based carrier screening
Carrier Testing • To determine an individual’s carrier status for a specific genetic disease • Not usually offered on a population basis
Carrier Testing • Available to clients with a family history of an autosomal recessive or X-linked genetic condition for which carrier testing available • e.g. Fragile X syndrome, Duchenne muscular dystrophy, Hemophilia A or B • e.g. PKU, Alpha-1-antitrypsin deficiency, Galactosemia
Ethnicity-Based Genetic Carrier Screening • Purpose: To detect couples at risk for prenatally diagnosable genetic diseases • Types of tests offered based on clients’ ethnic background • Offered to all individuals of that ethnic background (targeted population screening)
CARRIER FREQUENCIES BASED ON ETHNIC ORIGIN Condition Carrier Frequency Population
CF Carrier Results • Many tests detect a majority but not all carriers • Detection rates differ by ethnicity • Negative results do not eliminate risk • Different mutations may confer different risks • Example: CFTR R117H mutation and 5T allele • Genetic consultation is available to carriers and strongly advised for carrier/carrier couples
Drug Therapies • Medication are often aerosolize and can be inhaled • Bronchodilators • Mucolytics • Decongestants • Antibiotics to fight lung infections • Enzyme supplements
How can Gene Therapy be used to treat patients who suffer from Cystic Fibrosis (CF)?
Gene Therapy • SmaRT-discovered by Xiaoming Li • Interferes with the processing of RNA • Synthesis of DNA probe to locate the gene • make copies of normal and abnormal gene • Problems with gene therapy • Ethical issues • “Fooling with Mother Nature”
Gene Therapy Gene therapy is currently the most ambitious approach to curing Cystic Fibrosis Nondefective copies of the CFTR gene are introduced into affected cells, where they are taken up and used to create the CFTR protein This will result in the mucus functioning properly and a stop to Cystic Fibrosis
Gene Therapy It has been attempted to deliver the normal CFTR gene to the bronchial epithelium by aerosol spray, using a viral vector (usually an adenovirus) This viral vector would contain the genetic material for the CFTR gene The vector would be able to enter a cell inside the body and insert the genetic material for the CFTR gene into the cells DNA
Gene Therapy This would result in the cell having a normal CFTR gene and an end to CF Thus far the attempts have not been completely successful, as most patients develop an immune response against the virus during the course of therapy