Download

1 / 18
180 likes | 323 Views
PHM142 Fall 2013 Coordinator: Dr. Jeffrey Henderson Instructor: Dr. David Hampson. Cystic Fibrosis . Katalina Chan, Mary Vu, Sunny Wang and Vanessa Alexander Nov 4, 2013. Background. 80,000 CF patients Mutation in CFTR gene E pithelial cells at mucosal surfaces

E N D
PHM142 Fall 2013 Coordinator: Dr. Jeffrey Henderson Instructor: Dr. David Hampson Cystic Fibrosis Katalina Chan, Mary Vu, Sunny Wang and Vanessa Alexander Nov 4, 2013
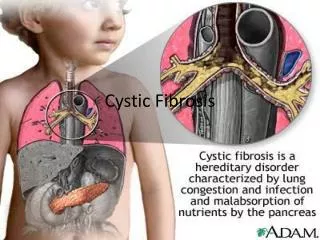
Background • 80,000 CF patients • Mutation in CFTR gene • Epithelial cells at mucosal surfaces • Viscid mucous secretions • Exocrine glands • Pancreas • Intestine • Liver • Bile duct
Background • Greatest mortality • Defect in airway mucociliary clearance • Early diagnosis • US median survival = 39 years
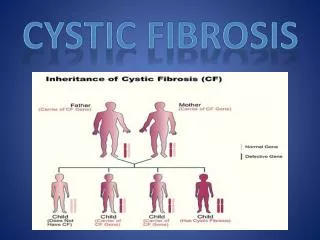
CF and Genetics • Caused by mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene • An autosomal recessive disease
CFTR Cystic fibrosis transmembrane conductance regulator protein Anion channel ATP Binding Cassette family Regulates other ion channels (ENaC) Plays crucial roles in absorption and secretion Found mainly in wet epithelia
CFTR Mutation and CF • Over 1000 mutations in the CFTR gene that can cause CF • Most common is ΔF508 • Abnormal biogenesis and premature degradation of the CFTR protein occurs • Results in defective ion transport across CF epithelia
Sweat Glands Pathophysiology • CFTR important for Cl- absorption • Na+ and Cl- generally reabsorbed • Cl- buildup = negative charge on luminal side • Decreased Na+ reabsorption • NaCl formation and secretion in sweat • Used as diagnostic factor
Rowe, S. M., Miller, S., & Sorscher, E. J. (2005). Cystic fibrosis. New England Journal of Medicine, 352(19), 1992-2002.
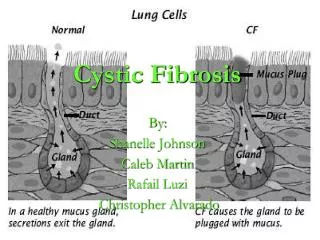
Lungs Pathophysiology • CFTR important for Cl- secretion • CFTR also mediate ENaC activity • Net decrease in Cl- secretion and increase in Na+ absorption • Osmotic surface mucous dehydrated • Buildup compresses cilia no mucous movement • Infections!
Clunes, M. T., & Boucher, R. C. (2007). Cystic fibrosis: The mechanisms of pathogenesis of an inherited lung disorder. NIH Drug Discovery Today, 4(2), 63-72.
Overall Pathophysiology • Sweat Glands hypersecretion of salt • Lungs mucous buildup + infection • Pancreas fibrous cysts + blockage • GI Tract blockage from thick feces • Liver obstruction, possible cirrhosis • Male Reproduction sterile
Airway Clearance Techniques • Mucolytics • Bronchodilators • The Vest • FLUTTER® • Exercise
Medication Antibiotics Other drugs • Correctors • Potentiators Example: inhaled Tobramycin P. aeruginosa
Nutrition • Pancreatic enzyme replacement therapy • 500-2500 lipase units/kg of body weight per meal • or 10,000 units/kg/day • or 4,000 units/g of dietary fat/day • Supplements for fat-soluble vitamins • Maintain healthy growth and weight • Males: BMI ≥ 23 • Females: BMI ≥ 22 • 110-200% of a healthy individual’s energy needs
Gene Therapy – Viral Vectors Problems: • Viral receptor is on basolateral side • Still trigger an immune response • Getting through the mucous http://www.pacificu.edu/optometry/ce/courses/20591/armdpg2.cfm
Lung Transplant • For those who are not expected to live 2 years • 5 year survival rate: ∼50%
Summary Slide • CF is caused by a mutation in the CFTR gene, affecting epithelial cells at mucosal surfaces (causes viscid mucous) • Symptoms: difficulty breathing, wet rattling cough, chronic lung infections, stunted growth, digestion issues • US median survival: 39 years • The CFTR protein regulates the transport of chloride and sodium ions across wet epithelial membranes. • The most common mutation (resulting in the loss of a phenylalanine in the protein) leads to defective ion transport. • Lack of CFTR leads to lack of chloride anion transport • In sweat glands, low chloride and sodium ion reabsorption = high salt secretion • In lungs, low chloride excretion, sodium ions hyper-absorbed into cells • Osmotic inbalance, dehydration of respiratory mucous = buildup which leads to infection and inflammation • Pathophysiology in most exocrine gland involves buildup in ducts which leads to blockage and infection • Airway clearance techniques – loosen mucous so it can be cleared • eg. mucolytics, bronchodilators, Flutter, Vest • Medication – antibiotics • Nutrition – PERT, vitamin supplements, high fat/protein diets • Gene therapy – viral vectors containing CFTR gene (a work in progress) • Lung transplant as a last resort
References • Clunes, M. T., & Boucher, R. C. (2007). Cystic fibrosis: The mechanisms of pathogenesis of an inherited lung disorder. NIH Drug Discovery Today, 4(2), 63-72. • Cuthbert, A. W. (2010). New horizons in the treatment of cystic fibrosis. British Journal of Pharmacology, 163, 173-183. • Cystic fibrosis Canada. (2009, July 30). Treatments for People with CF. Retrieved from http://www.cysticfibrosis.ca/en/treatment/Treatments.php • Cystic fibrosis Canada. (2011). Airway clearance techniques [PDF document]. Retrieved from http://www.cysticfibrosis.ca/assets/files/pdf/List_Airway_Clearance_TechniquesE.pdf • Dipiro, J. T., Talbert, R. L., Yee, G. C., Matzke, G. R., Wells, B. G., & Posey, L. M. (2011). Cystic fibrosis. Pharmacotherapy: A pathophysiologic approach (8th ed.). New York: McGraw-Hill. • Geller, D. E. & Rubin, B. K. (2009). Respiratory care and cystic fibrosis. Respiratory Care, 54(6), 796-800. • Hanrahan, J.W., Sampson, H.M., & Thomas, D.Y. (2013) Novel pharmacological strategies to treat cystic fibrosis. Trends in Pharmacological Sciences,32(2), 119-125. • Mueller, C. & Flotte, T. R. (2008). Gene therapy for cystic fibrosis. Clinical Reviews in Allergy & Immunology, 35(3), 164-178. • Ratjen, F. A. (2009). Cystic fibrosis: Pathogenesis and future treatment strategies. Respiratory Care, 54(5), 595-606. • Rowe, S. M., Miller, S., & Sorscher, E. J. (2005). Cystic fibrosis. New England Journal of Medicine, 352(19), 1992-2002. • Stallings, V. A., Stark, L.J., Robinson, K. A., Feranchak, A. P., & Quinton, H. (2008). Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: Results of a systematic review. Journal of the American Dietetic Association, 108(5), 832-839.