Download

1 / 98
1.01k likes | 1.55k Views
Amino Acid Metabolism. 生化教研室:牛永东. 生物化学的学习方法. 特点一:与数理化不同,尚未进入 定量科学 的阶段,还处在 定性科学 阶段。因此不可能通过公式或定理推出一个准确的结论; 特点二: 是没有绝对,几乎所有的结论都可以被一些例外打破(生物多样性)。 一般性结论:生物化学的学习应以概念为主 --- 以 记忆 为主,在记忆的基础上 加以理解 。. 【 目的与要求 】. 掌握脱氨基作用、 氨的来源、去路 ,氨的转运 …… 掌握尿素合成的部位和全过程 掌握 一碳单位 的概念、来源、载体和功能

E N D
Amino Acid Metabolism 生化教研室:牛永东
生物化学的学习方法 特点一:与数理化不同,尚未进入定量科学的阶段,还处在定性科学阶段。因此不可能通过公式或定理推出一个准确的结论; 特点二: 是没有绝对,几乎所有的结论都可以被一些例外打破(生物多样性)。 一般性结论:生物化学的学习应以概念为主---以记忆为主,在记忆的基础上加以理解。
【目的与要求】 • 掌握脱氨基作用、氨的来源、去路,氨的转运…… • 掌握尿素合成的部位和全过程 • 掌握一碳单位的概念、来源、载体和功能 • 需要掌握的概念: 必需氨基酸、蛋白质的互补作用、氨基酸库、联合脱氨基作用.……
Metabolism • consists of both catabolic and anabolic processes • Catabolism comprises all processes, in which complex molecules are broken down to simple ones • Anabolism means any constructive metabolic process by which organisms convert substances into other components required for the organism's chemical architecture
Introduction • Amino acids (AAs) are the building blocks of proteins (precursors for proteins) (物质代谢) • Energy metabolites (17.9KJ/g Pr):When degraded, amino acids produce glucose/carbohydrates and ketone bodies(能量代谢) • Precursors for many other biological N-containing compounds ,Involved as direct neurotransmitters or as precursors to neurotransmitters, eg. (信息分子代谢) • - Tyrosine gives DOPA and dopamine • - Precursors to peptide hormones and thyroid hormone • - Precursors to histamine, NAD and other compounds of biological importance
some major biological functions • Detoxification of drugs, chemicals and metabolic by-products • * Excess dietary AAs are neither storednor excreted. Rather, they are converted to common metabolic intermediates
outline 1. The nutrition of protein 2. The digestion、absorption and putrefaction of protein 3. The general metabolism of AA 4. Metabolism of ammonia 5. Single AA metabolism
Section 1 The nutrition of protein • Nitrogen balance • The requirements • Classification of amino acids
Nitrogen balance • Zero or total nitrogen balance: • the intake = the excretion (adult) • Amount of nitrogen intake is equal to the amount of nitrogen excreted is zero or total nitrogen balance • Positive nitrogen balance: • the intake > the excretion • during pregnancy, infancy, childhood and recovery from severe illness or surgery • Negative nitrogen balance: • the intake < the excretion • following severe trauma, surgery or infections. Prolonged periods of negative balance are dangerous and fatal if the loss of body protein reaches about one-third of the total body protein
The requirements • The requirements of protein for the health: the minimal requirement of protein is 30~50 gram for the adult • Advice: 80 gram/day (中国营养学会) ? ? ?
Classification of amino acids • non-essential amino acids • - can be synthesized by an organism • - usually are prepared from precursors in 1-2 steps • Essential amino acids *** • - cannot be made endogenously • - must be supplied in diet • eg. Leu, Phe…..
借来一两本淡色书 *The amino acidsArg, Hisare considered “conditionally essential” for reasons not directly related to lack of synthesis and they are essential for growth only
nutritional value • Legumes(豆类) poor in Trp, but rich in Lys; • Cereals (谷类) poor in Lys, but rich in Trp • Mutual complementation of amino acids • Protein deficiency-kwashiorkor, generalized edema and liver enlargement, abdomen bulged • Suggestion: the combined-action of protein in diet
Section 2The digestion、absorption and putrefaction of protein
Digestive Tract of protein • Proteins are generally too large to be absorbed by the intestine and therefore must be hydrolyzed to the amino acids • The proteolytic enzymes responsible for hydrolysis are produced by three different organs: the stomach、pancreas and small intestine (the major organ)
Stomach • HCl (parietal cells ) and Pepsinogen (chief cells ) • The pH of gastric juice is around 1.0. Food is retained in the stomach for 2-4 hrs • HCl kills microorganisms, denatures proteins, and provides an acid environment for the action of pepsin • Autocatalysis: pepsinogen is converted to active pepsin(Pepsin A) by HCl • Pepsin coagulates milk in presence of Ca2+ ions
Pancreas and small intestine • Endopeptidase(pancreas) • Trypsin:carbonyl of arg and lys • Chymotrypsin: carbonyl of Trp, Tyr, Phe, Met, Leu • Elastase: carbonyl of Ala, Gly, Ser • Exopeptidase(pancreas) • Carboxypeptidase A:amine side of Ala, Ile, Leu, Val • Carboxypeptidase B: amine side of Arg, lys • Aminopeptidase (small intestine): • cleaves N-terminal residue of oligopeptidaes • Dipeptidase (small intestine)
carboxypepidase endopeptidase aminopeptidase dipeptidase Amino acids+ 1/3 Amino acids 95%
absorption • There is little absorption from the stomach apart from short- and medium- chain fatty acids and ethanol • Under normal circumstances, the dietary proteins are almost completely digested to their constituent amino acids, and these end products of protein digestion are rapidly absorbed from the intestine into the portal blood
Amino acids are transported through the brush border by the carrier protein and it is an active transport • The classification of carrier protein: aciditic; basic; neutral and gly-carrier 2. -glutamyl cycle (-谷氨酰基循环) 3. The bi-and tri- peptidase carrier system in the intestinal mucosa cell
K+-ATPase Na+ Amino acids Carrier protein ATP Na+ Amino acids The mechanism of AA’s absorption K+ Na+ outer Member innner ADP+Pi K+ Na+ intestine
-Gln | | | | | | | | | | | | | | | | | | | | | | Cys-Gly peptidase -GGT ATP AA Cys Gly ADP+Pi Glu GSH ATP -glutamylcysteine ADP+Pi ATP -glutamyl cycle • -gltamyl cyclotransferase 5oxoprolinase membrane GCS synthetase ADP+Pi GSH synthetase
–CO2 Amines AA Putrefaction • Putrefaction: the process of decay of un-digestive and un-absorbed protein and the products by bacterial, fungal in the intestine 5% 1.Amines(胺): False neurotransmitter are similar with neurotransmitter
diffuseblood Ammonia intestine bacteria +NH3 AA 2. Ammonia(氨)-1: A. some amino acids are degraded by the in the intestine bacteria
urea CO2 +2NH3 diffuse blood Enter instein ammonia 2. Ammonia(氨)-2: B. urea from the blood to the intestine with resultant increased diffusion of NH3 into the intestinal Urea enzyme Urea in blood
3. The other toxic material: • phenol, indole, sulfureted hydrogen……
Section 4 The general metabolism of AA • Protein and amino acid turnover • Degradation of Amino Acids (Fate of amino group) • The metabolism of α-ketoacid
Body protein Reutilization for new protein synthesis Protein degradation Amino acids Protein turnover Protein and amino acid turnover 1-2% 75-80% T1/2 ? (half time)
introduction 1. Proteins constantly being synthesized and degraded - need constant supply of amino acids - need to degrade to protect from abnormal proteins - regulate cellular processes
2. Degraded by ubiquitin label - Ubiquitin binds lysine side chain - Targets for hydrolysis by proteosomes in cytosol and nucleus - ATP required 3. Degraded by the protease and the peptidase in the Lysosome - non- ATP required - the hydrolysis-selective are bad
E2-S- E1-S- The ubiquitin degradation pathway ATP AMP+PPi E3 E2-SH (ubiquitin) E1-SH E2-SH E1-SH E1:activiting enzyme E2:carrier protein E3:ligase ubiquitinational protein ATP 19S regulate substrate ATP 20S Proteasome 阿龙-西查诺瓦 26S Proteasome
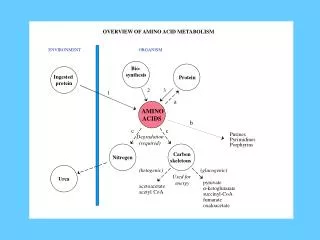
Diet protein Nonprotein nitrogen derivatives Tissue protein transamination Carbohydrate (glucose) Ketone dodies Acetyl-CoA Amino nitrogen in glutamate Citric Acid Cycle deamination NH3 Urea CO2 Amino acid pool Overview of the protein metabolism
- COO + a H N 3 H Degradation of Amino Acids - Reactions in amino acid metabolism Amino acid Carboxylic group Amino group R group
introduction • Free amino acids are metabolized in identical ways, regardless of whether they are released from dietary or intracellular proteins • The metabolism of the resulting amino group and nitrogen excretion are a central part of nitrogen metabolism
FATE OF AMINO GROUP DEAMINATION A. Transamination B. Oxidative deamination C. purine nucleotide cycle
A. Transamination • Transamination by Aminotransferase (transaminase) • always involve PLP coenzyme (pyridoxal phosphate) • reaction goes via a Schiff’s base intermediate • all transaminase reactions are reversible
Aminotransferases • Aminotransferases can have specificity for the alpha-keto acid or the amino acid • Aminotransferases exist for all amino acids except proline and lysine • The most common compounds involved as a donor/acceptor pair in transamination reactions are glutamate and a-ketoglutarate, which participate in reactions with many different aminotransferases • to an alpha-keto acid alpha-amino acid
Transamination aminotransferases
glutamate-pyruvate aminotransferase GPT, ALT Glu+pyruvate -Ketoglutarate+Ala (丙酮酸) (α-酮戊二酸) Glutamic oxaloacetictransaminase GOT, AST Glu+Oxaloacetate -Ketoglutarate+Asp (草酰乙酸) *** ALT and AST are components of a "liver function test". Levels increase with damage to liver (cirrhosis, hepatitis) or muscle (trauma)
–H2O + +H2O AA PLP Schiff’s base The mechanism of transamination
Molecule rearrange Schiff’s base isomer +H2O + –H2O -ketoacid PMP(磷酸吡哆胺)
Transamination • Interconversion of amino acids • Collection of N as glu • Provision of C-skeletons for catabolism
B. Oxidative Deamination • L-glutamate dehydrogenase (in mitochondria) • Glu + NAD+ (or NADP+) + H2O NH4+ + a-ketoglutarate + NAD(P)H +H+ • Requires NAD+ or NADP + as a cofactor • Plays a central role in AA metabolism ?
urea cycle ? It is inhibited by GTP and ATP, and activated by GDP and ADP
Combined deamination = Transamination + Oxidative Deamination The major pathway !!!
NH3 AA Asp IMP -Keto glutarate H2O AST aminotransferases C. purine nucleotide cycle AMP -Keto acid Oxaloacetate fumarate malate
The metabolism of α-ketoacid • Biosynthesis of nonessential amino acids • TCA cycle member + amino acid α-keto acid + nonessential amino acid • A source of energy (10%) ( CO2+H2O ) • Glucogenesis and ketogenesis
* Classification of amino acids • * glucogenic amino acid : are converted into either pyruvate or one of the citric acid cycle intermediates • (a-ketoglutarate, succinyl CoA, fumarate or malate) • * ketogenic amino acid: will be deaminated via Acetylc-CoA and thus can be made into a ketone body. such as: Leucine and lysine • * glucogenic and ketogenic amino acid: isoleucine, phenylalanine, tryptophan and tyrosine, threonine