Download

1 / 60
600 likes | 783 Views
Alignment of Flexible Protein Structures. Based on: FlexProt: Alignment of Flexible Protein Structures Without a Pre-definition of Hinge Regions / M. Shatsky, R. Nussinov, H. Wolfson. Presented by: Einat Engel. Introduction. Proteins are flexible structures. Outline. Introduction

E N D
Alignment of Flexible Protein Structures Based on: FlexProt: Alignment of Flexible Protein Structures Without a Pre-definition of Hinge Regions / M. Shatsky, R. Nussinov, H. Wolfson Presented by: Einat Engel
Introduction Proteins are flexible structures
Outline Introduction • Proteins (reminder) • Protein motion • Structural alignment – rigid & flexible General Description: • Problem’s description • Discussion
Outline Detailed Description: • FPSA problem description • FlexProt algorithm for the FPSA problem • Experimental results • Heuristic Algorithm for FPSA • Clustering Conclusions & Discussion: • Summary of algorithm • Major results • Discussion
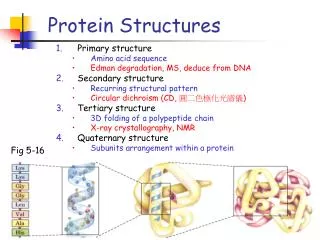
Reminder: Protein Structure Proteins are made up of 20 different amino acids (or "residues"). Different levels of protein structures: • Primary – amino acid sequence • Secondary – local folding of amino acid chains • Tertiary – 3D structure of a protein • Quaternary – forming multi-chained proteins
Reminder: Protein Structure Primary structure Tertiary structure lysozyme
Flexibility & Protein Motion • Proteins are flexible molecules that undergo significant structural changes as part of their normal function. • Motion often serves as an essential link between structure and function.
Flexibility & Protein Motion • Protein motions are involved in numerous basic functions. In fact, highly mobile proteins have been implicated in a number of diseases, e.g., the motion of gp41 in AIDS
Structural Alignment • When flexible molecules are compared to each other as rigid bodies, even strong similarities can be missed • Yet, most existing protein alignment algorithms treat them as rigid objects • We’ll see a technique for the alignment of flexible proteins
The Goal Go back
Existing Approaches – Rigid Structural Alignment • Exhaustive 3D search – search all possible rotations. (Matthews & Rossman) • Fragment alignment – comparison of contiguous fragments. • Geometric Hashing – Local reference frame, preprocessing & recognition (Fischer) • Curve Matching – match curves using Fourier Transform (Schwartz & Sharir)
Existing Approaches – Flexible Structural Alignment • Domain detection – requires a-priori knowledge of the corresponding pairs of amino-acid residues (Wriggers & Schulten) • Geometric hashing – requires a-priori knowledge of the hinge location (Verbitsky) • Data base screening – requires a-priori knowledge of hinges (Rigoutsos)
Outline Introduction • Proteins (reminder) • Protein motion • Structural alignment – rigid & flexible General Description: • Problem’s description • Discussion
Terminology Two fragments are almost congruent (matched) if: • Their sequence length is the same. • There exists a 3D rotation and translation which superimposes the corresponding atoms with small RMSD. (Reminder: RMSD measures alignment error.)
Problem Definition • Input: two protein molecules M1 and M2. • Task: divide the two molecules into fragments of maximal size, such that the matched fragments will be almost congruent.
Problem Discussion • The regions between the fragments are called flexible (hinge) regions. • We’d like to minimize the number of flexible regions and maximize the alignment size • Our goal is to find a balanced solution Example Conflict!
Problem Discussion Consider two different solutions: I. 3 rigid parts. Total size = 200 atoms II. 2 rigid parts. Total size = 150 atoms Q:Which is better? A: I don’t know. Let’s divide the results according to the number of rigid parts.
Major Results • Introducing FlexProt, a new technique for the alignment of flexible proteins. • Unlike other algorithms, FlexProt does not require a priori knowledge of the locations of the flexible, hinge-bending sites • The pairs of rigid matching fragments and the flexible regions are detected simultaneously
Outline Detailed Description: • FPSA problem description • FlexProt algorithm for the FPSA problem • Experimental results • Heuristic Algorithms for FPSA • Clustering Conclusions & Discussion: • Summary of algorithm • Major results • Discussion
Flexible Protein Structural Alignment (FPSA) • Input • Two proteins, • Threshold error MaxRMSD • MaxFlexNum parameter • A weight function w
and has the following property: A rigid fragment pair is defined as: Where is defined as follows: FPSA Problem Terminology T is a 3D rigid transformation, meaning rotation and translation
Let be a list of rigid fragment pairs where , such that Let FPSA Problem Terminology w is a weight function that reflects the “goodness” of linking two rigid fragment pairs.
The FPSA Problem Example:
For Each detect such that: Remember, is a list of rigid fragment pairs The FPSA Problem
The FlexProt Algorithm for FPSA I. Detection of all rigid fragment pairs, that satisfy the MaxRMSD constraint II. Detection of optimal configurations between rigid fragment pairs,
Iterate over three indices where I. Detect all Rigid Fragment Pairs In order to find all possible pairs, , do: and select the pairs satisfying
We assume that • Iterate over I. Complexity Remember, a rigid fragment pair - • Compute RMSD for each triplet – linear in the detected fragment size (Sharir) Total complexity -
II. Detect Optimal Configuration • Now, we have a set of congruent fragment pairs. • Let’s find an optimal subset of it. This subset will describe an alignment of M2with M1. We’ll use dynamic programming Dynamic programming – solves optimization problem by caching subproblem solutions rather than recomputing them. Dynamic programming – solves optimization problem by caching subproblem solutions rather than recomputing them.
II. Detect Optimal Configuration • In General: define a graph • Vertices represent the rigid fragment pairs • The directed edges represent flexible regions connecting the rigid fragment pairs • A weight function w is applied to the edges. it reflects the goodness of connecting two rigidly matched fragment pairs
Vertices II. Detect Optimal Configuration A directed edge between and is defined if: 1. The fragments are ascending 2. The gaps between consecutive fragments are limited by MaxGap1and MaxGap2(user defined)
II. Detect Optimal Configuration Define: MaxGap1=3 MaxGap2=3 C A B
II. Detect Optimal Configuration The weight function (smaller is better): Δis half of the maximal overlapping interval • Part A rewards quadratically the size of • Part B punishes large gaps • Part C punishes difference between Gap1and Gap2
II. Detect Optimal Configuration C e2 e1 A B
II. Detect Optimal Configuration • We built a weighted directed acyclic graph (DAG) • Shortest weighted paths correspond to alignments of consecutive, long, congruent matching fragments. Almost Finished
Reminder: Shortest Paths in DAGs First, we perform a topological sort of the Directed Acyclic Graph (DAG). Then, we make just one pass over the vertices according to their order. For each vertex, we relax each edge that leaves it. 1 6 0 0 2 ∞ 2 2 6 6 ∞ ∞ 5 5 6 4 3 3 ∞ 7 -1 -2 4 2
II. Detect Optimal Configuration • We run the Shortest Paths in DAGs algorithm. • A simple case (no limit on the number of nodes in the shortest path): The shortest path in G corresponds to a minimal weighted sequence of rigid fragment pairs, F**, such that • Complexity -
II. Detect Optimal Configuration • We’ll make a small change in the algorithm since we need to detect shortest paths with exactly s nodes, • In the simple case, each node holds a pointer to a preceding node with the shortest path. • Instead, each node will hold MaxFlexNum pointers. Pointer s points to a preceding node with a shortest path of size s-1
The number of nodes in the graph can be proportional to • Graph of n vertices has edges Total complexity of stage II: II. Complexity During Relaxation, we check all MaxFlexNum possibilities and therefore the complexity is
Summary of FlexProt Algorithm • Theoretical worst case complexity is • In practice – FlexProt is highly efficient (with some changes) • The average running time is approximately seven seconds (for molecules of 300 amino acids) So… What does it look like??
Experimental Results Experimental Results
Running FlexProt • http://bioinfo3d.math.tau.ac.il/FlexProt • http://www.umass.edu/microbio/chime/explorer/pe.htm
Heuristic Improvement of Step I • In step I, we detected all of the rigid fragment pairs. Time complexity – • The procedure takes several minutes, even for small proteins. • Instead, we can use a greedy algorithm, that only takes
Heuristic Improvement of Step I • Start by aligning a single matching atom pair where and • Iteratively, add one matching atom pair to the left and one to the right. • Stop when we exceed the RMSD threshold – when the list can’t be extended to the left or the right.
is almost congruent to The next alignment is a-1 j+l-1 a+1 initiated at i+l-1 a b-1 j b+1 and not b i Heuristic Improvement of Step I After the extension process, we have a match-list
Thus, finding a particular is linear in the length of the fragments - The time complexity, is: Complexity Updating the RMSD at each step is
Theoretically, some atom pairs , might participate in at most n fragment pairs. In practice, a pair of atoms participates in at most 2 fragment pairs. Complexity There are O(n2) rigid fragment pairs
Clustering • This stage can be viewed as an extension to the FPSA problem. • The algorithm clusters consecutive fragment pairs, that have a similar 3D transformation, even if they are not directly linked.
Clustering Example: Two β-strands (A and B) are connected by loops of different lengths. Stage I of the FlexProt algorithm aligns each separately. A and B have almost the same 3D rigid transformation and in the clustering stage, they are joined into one structure.
The Clustering Algorithm • We take each path detected in stage II. Remember, vertices = congruent fragment pair • The first vertex is a singleton cluster • Take the second vertex. Check if there is a rigid transformation which superimposes both fragments. If successful – do the same for the next vertex Else – start a new cluster with the vertex that failed to join the previous cluster