Download
1 / 41
431 likes | 537 Views
Learn about using restriction enzymes like EcoRI for gene manipulation, plasmid vectors, PCR, cDNA synthesis, and gene cloning methods. Explore recombinant DNA technology and applications in biotechnology.

E N D
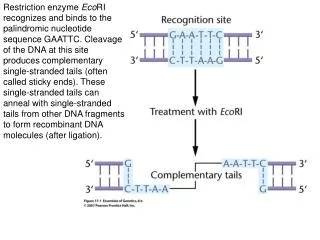
Restriction enzyme EcoRI recognizes and binds to the palindromic nucleotide sequence GAATTC. Cleavage of the DNA at this site produces complementary single-stranded tails (often called sticky ends). These single-stranded tails can anneal with single-stranded tails from other DNA fragments to form recombinant DNA molecules (after ligation).
DNA from different sources is cleaved with EcoRI and mixed to allow annealing. The enzyme DNA ligase then chemically bonds these annealed fragments into an intact recombinant DNA molecule.
This genome contains over 48.5 Kbp DNA. The molecule has been spread under partial denaturing conditions in order to see the unwound A+T rich regions which are located on the right half of this genome
Circular plasmid molecules isolated from E. coli. Genetically engineered plasmids are used as vectors for cloning DNA.
Plasmid pUC18 showing the polylinker region, located within a lacZ gene. DNA inserted into the polylinker region disrupts the lacZ gene, resulting in white colonies that allow direct identification of bacterial colonies carrying cloned DNA inserts
A Petri dish showing the growth of bacterial cells after uptake of recombinant plasmids. The medium on the plate contains a compound called Xgal. DNA cloned into the pUC 18 vector disrupts the gene responsible for metabolism of Xgal and formation of blue colonies. As a result, it is easy to distinguish colonies carrying cloned DNA inserts. Cells in blue colonies contain vectors without cloned DNA inserts, whereas cells in white colonies contain vectors carrying DNA inserts.
The plasmid vector pUC18 carries unique restriction enzyme cleavage sites in a polylinker region within the lacZ gene. Cleavage of the plasmid and DNA to be cloned with a restriction enzyme and insertion of a DNA fragment into the polylinker region disrupts the lacZ gene, making plasmids carrying inserts unable to metabolize Xgal, resulting in the formation of white colonies on nutrient plates containing Xgal.
The yeast artificial chromosome pYAC3 contains telomere sequences (TEL), a centromere (CEN4) derived from yeast chromosome 4, and an origin of replication (ori). These elements give the cloning vector the properties of a chromosome. TRP1 and URA3 are yeast genes that are selectable markers for the left and right arms of the chromosome. Within the SUP4 gene is a restriction site for the enzyme SnaB1. Two BamH1 restriction sites flank a spacer segment. Cleavage with SnaB1 and BamH1 breaks the artificial chromosome into two arms. The DNA to be cloned is treated with SnaB1, producing a collection of fragments. The arms and fragments are ligated together, and the artificial chromosome is inserted into yeast host cells. Because yeast chromosomes are large, the artificial chromosome accepts inserts in the million base-pair range
Cloning with a plasmid vector involves cutting both plasmid and the DNA to be cloned with the same restriction enzyme. The DNA to be cloned is spliced into the vector and transferred to a bacterial host for replication. Bacterial cells carrying plasmids with DNA inserts can be identified by selection or screening and then isolated. The cloned DNA is then recovered from the bacterial host for further analysis.
In the polymerase chain reaction (PCR), the target DNA is denatured into single strands; each strand is then annealed to a short, complementary primer. DNA polymerase extends the primers in the 3' direction, using the single-stranded DNA as a template. The result is two newly synthesized double-stranded DNA molecules with the primers incorporated into them. Repeated cycles of PCR can amplify the original DNA sequence by more than a millionfold.
Producing cDNA from mRNA. Because many eukaryotic mRNAs have a polyadenylated tail (A) of variable length at their 3' end, a short oligo-dT annealed to this tail serves as a primer for the enzyme reverse transcriptase. Reverse transcriptase uses the mRNA as a template to synthesize a complementary DNA strand (cDNA) and forms an mRNA/cDNA double-stranded duplex. The mRNA is digested with the enzyme RNAse H, producing gaps in the RNA strand. The 3' ends of the remaining RNA serve as primers for DNA polymerase, which synthesizes a second DNA strand. The result is a double-stranded cDNA molecule that can be cloned into a suitable vector or used directly as a probe for library screening.
Screening a plasmid library to recover a cloned gene. The library, present in bacteria on Petri plates, is overlaid with a DNA binding filter, and colonies are transferred to the filter. Colonies on the filter are lysed, and the DNA is denatured to single strands. The filter is placed in a hybridization bag along with buffer and a labeled single-stranded DNA probe. During incubation, the probe forms a double-stranded hybrid with complementary sequences on the filter. The filter is removed from the bag and washed to remove excess probe. Hybrids are detected by placing a piece of X-ray film over the filter and exposing it for a short time. The film is developed, and hybridization events are visualized as spots on the film. Colonies containing the insert that hybridized to the probe are identified from the orientation of the spots. Cells are picked from this colony for growth and further analysis.
Constructing a restriction map. Samples of the 7.0 kb DNA fragments are digested with restriction enzymes: One sample is digested with HindIII, one with SalI, and one with both HindIII and SalI. The resulting fragments are separated by gel electrophoresis. The sizes of the separated fragments are measured by comparing them with molecular-weight standards in an adjacent lane. Cutting the DNA with HindIII generates two fragments: 0.8 kb and 6.2 kb. Cutting with SalI produces two fragments: 1.2 kb and 5.8 kb. Models are constructed to predict the fragment sizes generated by cutting with both HindIII and SalI. Model 1 predicts that 0.4, 0.8, and 5.8 kb fragments will result from cutting with both enzymes. Model 2 predicts that 0.8, 1.2, and 5.0 kb fragments will result. Comparing the predicted fragments with those observed on the gel indicates that model 1 is the correct restriction map.
Southern blotting technique: samples of the DNA to be probed are cut with restriction enzymes and the fragments are separated by gel electrophoresis. The pattern of fragments is visualized and photographed under ultraviolet illumination by staining the gel with ethidium bromide. The gel is then placed on a sponge wick in contact with a buffer solution and covered with a DNA-binding filter. Layers of paper towels or blotting paper are placed on top of the filter and held in place with a weight. Capillary action draws the buffer through the gel, transferring the pattern of DNA fragments from the gel to the filter. The DNA fragments on the filter are then denatured into single strands and hybridized with a labeled DNA probe. The filter is washed to remove excess probe and overlaid with a piece of X-ray film for autoradiography. The hybridized fragments show as bands on the X-ray film.
(a) Agarose gel stained with ethidium bromide to show DNA fragments. (b) Exposed X-ray film of a Southern blot prepared from the gel in part (a). Only those bands containing DNA sequences complementary to the probe show hybridization.
Deoxynucleotides (top) have an OH group at the 3' position in the ribose molecule. Dideoxynucleotides (bottom) lack an OH group at this position, preventing formation of a phosphodiester bond with another nucleotide, terminating further elongation of the template strand.
DNA sequencing gel showing the separation of newly synthesized fragments in the four sequencing reactions (one per lane). To obtain the base sequence of the DNA fragment, the gel is read from the bottom, beginning with the lowest band in any lane, then the next lowest, then the next, and so on. For example, the sequence of the DNA on this gel begins with 5'-CG at the very bottom of the gel, proceeds upwards as 5'-CGCTTTCATGTCA, and so forth.
In DNA sequencing using dideoxynucleotides labeled with fluorescent dyes, all four ddNTPs are added to the same tube, and during primer extension, all combinations of chains are produced. The products of the reaction are added to a single lane on a gel, and the bands are read by a detector and imaging system. This process is now automated, and robotic machines, such as those used in the Human Genome Project, sequence several hundred thousand nucleotides in a 24-hour period and then store and analyze the data automatically. The sequence obtained is by extension of the primer and is read from the newly synthesized strand, not the template strand. Thus, the sequence obtained begins with 5'-CTAGACATG.
Automated DNA sequencing using fluorescent dyes, one for each base. Each peak represents the correct nucleotide in the sequence. The sequence extending from the primer (which is not shown here) starts at the upper left of the diagram and extends rightward. The bases labeled as N are ambiguous and cannot be identified with certainty. These ambiguous base readings are more likely to occur near the primer because the quality of sequence determination deteriorates the closer the sequence is to the primer. The separated bases are read in order along the axis from left to right. Thus, the sequence begins as 5'-TGNNANACTGACNCAC. Numbers below the bases indicate length of the sequence in base pairs.