Enhancing Bioinformatics with SAT: Haplotype Inference and Consistency Checking
10 likes | 141 Views
This presentation explores the applications of SAT (Satisfiability) and Pseudo-Boolean optimization in bioinformatics. We discuss methods for haplotype inference from a set of genotypes, ensuring that every genotype can be explained by pairs of haplotypes. In addition, we examine pedigree consistency checking according to Mendelian inheritance laws. We also cover protein folding and genomic distance calculations, highlighting efficient algorithms and compact models provided by SAT and its extensions. Insights into future approaches for optimization and symmetry reduction are also presented.

Enhancing Bioinformatics with SAT: Haplotype Inference and Consistency Checking
E N D
Presentation Transcript
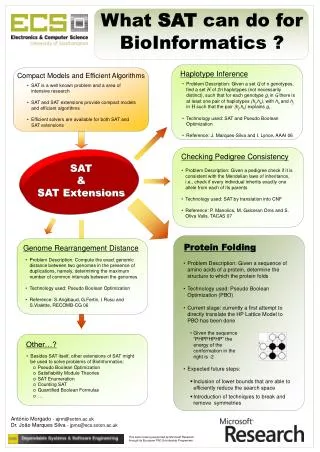
What SAT can do for BioInformatics ? Haplotype Inference Compact Models and Efficient Algorithms • Problem Description: Given a setGof n genotypes, find a setHof 2n haplotypes (not necessarily distinct), such that for each genotype gi in Gthere is at least one pair of haplotypes (hj,hk), with hkand hj inHsuch that the pair (hj,hk) explains gi • Technology used: SAT and Pseudo Boolean Optimization • Reference: J. Marques-Silva and I. Lynce, AAAI 06 • SAT is a well known problem and a area of intensive research • SAT and SAT extensions provide compact models and efficient algorithms • Efficient solvers are available for both SAT and SAT extensions Checking Pedigree Consistency SAT & SAT Extensions • Problem Description: Given a pedigree check if it is consistent with the Mendelian laws of inheritance, i.e., check if every individual inherits exactly one allele from each of its parents • Technology used: SAT by translation into CNF • Reference: P. Manolios, M. Galceran Oms and S. Oliva Valls, TACAS 07 Protein Folding Genome Rearrangement Distance • Problem Description: Compute the exact genomic distance between two genomes in the presence of duplications, namely, determining the maximum number of common intervals between the genomes • Technology used: Pseudo Boolean Optimization • Reference: S.Angibaud, G.Fertin, I.Rusu and S.Vialette, RECOMB-CG 06 • Problem Description: Given a sequence of amino acids of a protein, determine the structure to which the protein folds • Technology used: Pseudo Boolean Optimization (PBO) • Current stage: currently a first attempt to directly translate the HP Lattice Model to PBO has been done • Given the sequence • “PHPPHPHP” the • energy of the • conformation in the • right is -2 • Expected future steps: • Inclusion of lower bounds that are able to efficiently reduce the search space • Introduction of techniques to break and remove symmetries Other…? • Besides SAT itself, other extensions of SAT might be used to solve problems of BioInformatics: • Pseudo Boolean Optimization • Satisfiability Module Theories • SAT Enumeration • Counting SAT • Quantified Boolean Formulas • … António Morgado - ajrm@soton.ac.uk Dr. João Marques Silva - jpms@ecs.soton.ac.uk This work is being supported by Microsoft Research through its European PhD Scholarship Programme




















