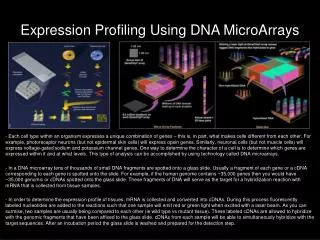
DNA Microarrays
DNA Microarrays. Patrick Schmid CSE 497 Spring 2004. What is a DNA Microarray?. Also known as DNA Chip Allows simultaneous measurement of the level of transcription for every gene in a genome (gene expression) Transcription? Process of copying of DNA into messenger RNA (mRNA)


DNA Microarrays
E N D
Presentation Transcript
DNA Microarrays Patrick Schmid CSE 497 Spring 2004
What is a DNA Microarray? • Also known as DNA Chip • Allows simultaneous measurement of the level of transcription for every gene in a genome (gene expression) • Transcription? • Process of copying of DNA into messenger RNA (mRNA) • Environment dependant! • Microarray detects mRNA, or rather the more stable cDNA Patrick Schmid
What is a DNA Microarray? (cont.) Cheung et al. 1999 Patrick Schmid
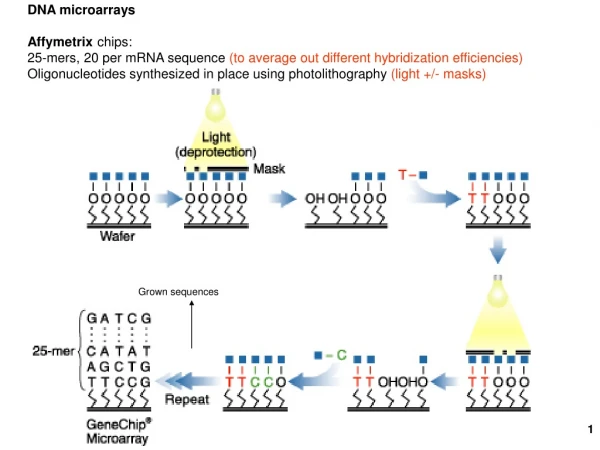
How do we manufacture a microarray? • Start with individual genes, e.g. the ~6,200 genes of the yeast genome • Amplify all of them using polymerase chain reaction (PCR) • “Spot” them on a medium, e.g. an ordinary glass microscope slide • Each spot is about 100 µm in diameter • Spotting is done by a robot • Complex and potentially expensive task Patrick Schmid
How do we manufacture a microarray? Cheung et al. 1999 Patrick Schmid
Example • Remember the flash animation? • Yeast • Grow in aerobic and anaerobic environment • Different genes will be activated in order to adapt to each environment • Extract mRNA • Convert mRNA into colored cDNA (fluorescently labeled) Patrick Schmid
Example (cont.) • Mix cDNA together • Hybridize cDNA with array • Each cDNA sequence hybridizes specifically with the corresponding gene sequence in the array • Wash unhybridized cDNA off • Read array with laser • Analyze images Patrick Schmid
Overview of Example Brown & Botstein, 1999 Patrick Schmid
Reading an array • Laser scans array and produces images • One laser for each color, e.g. one for green, one for red • Image analysis, main tasks: • Noise suppression • Spot localization and detection, including the extraction of the background intensity, the spot position, and the spot boundary and size • Data quantification and quality assessment • Image Analysis is a book on its own: • Kamberova, G. & Shah, S. “DNA Array Image Analysis Nuts & Bolts“. DNA Press LLC, 2002 Patrick Schmid
Reading an array (cont.) Campbell & Heyer, 2003 Patrick Schmid
Real DNA Microarray Campbell & Heyer, 2003 Patrick Schmid
Y-fold • Biologists rather deal with folds than with ratios • A fold is nothing else than saying “times” • We express it either as a Y-fold repression, or a Y-fold induction • It is calculated by taking the inverse of the ratio • Ratio of 0.33 = 3-fold repression • Ratio of 10 = 10-fold induction • Fractional ratios can cause problems with techniques of analyzing and comparing gene expression patterns Patrick Schmid
Color Coding • Tables are difficult to read • Data is presented with a color scale • Coding scheme: • Green = repressed (less mRNA) gene in experiment • Red = induced (more mRNA) gene in experiment • Black = no change (1:1 ratio) • Or • Green = control condition (e.g. aerobic) • Red = experimental condition (e.g. anaerobic) • We only use ratio Campbell & Heyer, 2003 Patrick Schmid
Logarithmic transformation • log2 is commonly used • Sometimes log10 is used • Example: • log2(0.0625) = log2(1/16) = log2(1) – log2(16) = -log2(16) = -4 • log2 transformations ease identification of doublings or halvings in ratios • log10 transformations ease identification of order of magnitude changes • Key attribute: equally sized induction and repression receive equal treatment visually and mathematically Patrick Schmid
Complication: Time Series • Biologists care more about the process of adaptation than about the end result • For example, measure every 2 hours for 10 hours (depletion of oxygen) • 31,000 gene expression ratios • Or 6,200 different graphs with five data points each • Question: Are there any genes that responded in similar ways to the depletion of oxygen? Patrick Schmid
Example data: fold change (ratios) What is the pattern? Campbell & Heyer, 2003 Patrick Schmid
Example data: log2 transformation Campbell & Heyer, 2003 Patrick Schmid
Pearson Correlation Coefficient r • Gene expression over time is a vector, e.g. for gene C: (0, 3, 3.58, 4, 3.58, 3) • Given two vectors X and Y that contain N elements, we calculate r as follows: Cho & Won, 2003 Patrick Schmid
Pearson Correlation Coefficient r (cont.) • X = Gene C = (0, 3.00, 3.58, 4, 3.58, 3)Y = Gene D = (0, 1.58, 2.00, 2, 1.58, 1) • ∑XY = (0)(0)+(3)(1.58)+(3.58)(2)+(4)(2)+(3.58)(1.58)+(3)(1) = 28.5564 • ∑X = 3+3.58+4+3.58+3 = 17.16 • ∑X2 = 32+3.582+42+3.582+32 = 59.6328 • ∑Y = 1.58+2+2+1.58+1 = 8.16 • ∑Y2 = 1.582+22+22+1.582+12 = 13.9928 • N = 6 • ∑XY – ∑X∑Y/N = 28.5564 – (17.16)(8.16)/6 = 5.2188 • ∑X2 – (∑X)2/N = 59.6328 – (17.16)2/6 = 10.5552 • ∑Y2 – (∑Y)2/N = 13.9928 – (8.16)2/6 = 2.8952 • r = 5.2188 / sqrt((10.5552)(2.8952)) = 0.944 Patrick Schmid
Example data: Pearson correlation coefficient Campbell & Heyer, 2003 Patrick Schmid
Example: Reorganization of data Campbell & Heyer, 2003 Patrick Schmid
Clustering of example Campbell & Heyer, 2003 Patrick Schmid
Clustering of entire yeast genome Campbell & Heyer, 2003 Patrick Schmid
Hierarchical Clustering • Algorithm: • First, find the two most similar genes in the entire set of genes. Join these together into a cluster. Now join the next two most similar objects (an object can be a gene or a cluster), forming a new cluster. Add the new cluster to the list of available objects, and remove the two objects used to form the new cluster. Continue this process, joining objects in the order of their similarity to one another, until there is only one object on the list – a single cluster containing all genes.(Campbell & Heyer, 2003) Patrick Schmid
Hierarchical Clustering (cont.) Campbell & Heyer, 2003 Patrick Schmid
Hierarchical Clustering (cont.) C D E • Average observations • Gene D: (0.94+0.84)/2 = 0.89 • Gene F: (-0.40+(-0.57))/2 = -0.485 • Gene G: (0.95+0.89)/2 = 0.92 F 1 G C E Patrick Schmid
Hierarchical Clustering (cont.) D F 1 2 G C E G D Patrick Schmid
Hierarchical Clustering (cont.) 3 F 1 2 C E G D Patrick Schmid
Hierarchical Clustering (cont.) 4 3 F 1 2 F C E G D Patrick Schmid
Hierarchical Clustering (cont.) Did this algorithm not look familiar? 4 Remember Neighbor-Joining? 3 1 2 F C E G D Patrick Schmid
Hierarchical Clustering (cont.) Eisen et al., 1998 Patrick Schmid
Hierarchical Clustering (cont.) • We differentiate hierarchical clustering algorithms by how they agglomerate distances: • Single Linkage • Shortest link between two clusters • Complete Linkage • Longest link between two clusters • Average Linkage • Average of distances between all pairs of objects • Average Group Linkage • Groups once formed are represented by their mean values, and then those are averaged • Which one did we use in the previous example ? http://www.resample.com/xlminer/help/HClst/HClst_intro.htm Patrick Schmid
Clustering Overview • Different similarity measures • Pearson Correlation Coefficient • Cosine Coefficient • Euclidean Distance • Information Gain • Mutual Information • Signal to noise ratio • Simple Matching for Nominals Patrick Schmid
Clustering Overview (cont.) • Different Clustering Methods • Unsupervised • Hierarchical Clustering • k-means Clustering (k nearest neighbors) • Thursday • Self-organizing map • Thursday • Supervised • Support vector machine • Ensemble classifier • Data Mining Patrick Schmid
Support Vector Machines • Linear regression: • x = w0 + w1a1 + w2a2 + … + wkak • x is the class, ai are the attribute values and wj are the weights • Given a distance vector Y with distances ai in which class x does Y belong? • What do we mean by a class x? • Primitive method: Y is in one class if x<0.5, in another class for x≥0.5. Patrick Schmid
Support Vector Machines (cont.) • Multi-response linear regression: • Set output to 1 for training instances that belong to a class • Set output to 0 for training instances that do not belong to that class • Result is a linear expression for each class • Classification of unknown example: • Compute all linear expressions • Choose the one that gives the largest output value Patrick Schmid
Support Vector Machines (cont.) • This means… • Two pairs of classes • Weight vector for class 1: • w0(1) + w1(1)a1 + w2(1)a2 + … + wk(1)ak • Weight vector for class 2: • w0(2) + w1(2)a1 + w2(2)a2 + … + wk(2)ak • An instance will be assigned to class 1 rather than class 2 if • w0(1) + w1(1)a1 + w2(1)a2 + … + wk(1)ak> w0(2) + w1(2)a1 + w2(2)a2 + … + wk(2)ak • We can rewrite this as • (w0(1) - w0(2)) + (w1(1) -w1(2)) a1 + … + (wk(1) - wk(2)) ak > 0 • Hyperplane Patrick Schmid
Support Vector Machines (cont.) • We can only represent linear boundaries between classes so far • Trick: Transform the input using a nonlinear mapping, then construct a linear model in the new space • Example: Use all products of n factors (2 attributes, n=3): • x = w1a13 + w2a12a2 + w3a1a22 + w4a23 • Then use multi-response linear regression • However, for 10 attributes and including all products with 5 factors, we would need to determine more than 2000 coefficients • Linear regression is O(n3) in time • Problem: Training is infeasible • Another problem: Overfit. The resulting model will be “too nonlinear”, because there are just too many parameters in the model. Patrick Schmid
Support Vector Machines (cont.) • Convex hull of points is the tightest enclosing polygon • Maximum margin hyperplane • Instances closest to hyperplane are called support vectors • Support vectors define maximum margin hyperplane uniquely support vectors Witten & Frank, 2000 Patrick Schmid
Support Vector Machines (cont.) • We only need set of support vectors, everything else is irrelevant • A hyperplane separating two classes can then be written as • x = w0 + w1a1 + w2a2 • Or • x = b + ∑ αiγi (a(i) ∙ a) • i is support vector • γi is the class value of a(i) • b and αi are numeric values to be determined • Vector a represents a test instance • a(i) are the support vectors • Determining b and αi is a constrained quadratic optimization problem that can be solved with off-the-shelf software packages • Support Vector Machines do not overfit, because there are usually only a few support vectors Patrick Schmid
Support Vector Machines (cont.) • Did I not introduce Support Vector Machines by talking about non-linear class boundaries? • x = b + ∑ αiγi (a(i) ∙ a)n • n is the number of factors • (x ∙ y)n is called a polynomial kernel • A good way of choosing n is by starting with n=1 and incrementing it until estimated error ceases to improve • If you want to know more: • SVMs in general: Witten & Frank, 2000 (lecture material based on this) • Application to cancer classification: Cho & Won, 2003 Patrick Schmid
References • Brown, P., Botstein, D. “Exploring the new world of the genome with DNA microarrays” Nature genetics supplement, vol. 21, January 1999 • Campbell A. & Heyer L. “discovering Genomics, Proteomics, & Bioninformatics” Benjamin Cummings, 2003. • Cheung, V., Morley, M., Aguilar, F., Massimi, A., Kucherlapati, R. & Childs, G. “Making and reading microarrays” Nature genetics supplement, vol. 21, January 1999 • Cho, S. & Won, H. “Machine Learning in DNA Microarray Analysis for Cancer Classification” Proceedings of the First Asia-Pacific bioinformatics conference on Bioinformatics 2003 - Volume 19, Australian Computer Society Inc. • Eisen, M., Spellman, P., Brown, P. & Botstein, D. “Cluster analysis and display of genome-wide expression patterns” Proc. Natl. Acad. Sci. USA. Vol 95, pp. 14 863-14868, December 1998. Genetics • Seo, J. & Sheiderman, B. “Interactively Exploring Hierarchical Clustering Results” IEEE Computer, July 2002 • Witten, I. & Frank, E. “Data Mining” Morgan Kaufmann Publishers, 2000 Patrick Schmid