
Basic Local Alignment Search Tool (BLAST) Program Query Database
Basic Local Alignment Search Tool (BLAST) Program Query Database --------------------------------------------------------------------------------- BLASTN Nucleotide Nucleotide BLASTP Protein Protein BLASTX Nucleotide=Protein Protein

Basic Local Alignment Search Tool (BLAST) Program Query Database
E N D
Presentation Transcript
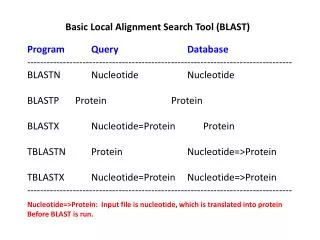
Basic Local Alignment Search Tool (BLAST) Program Query Database --------------------------------------------------------------------------------- BLASTN Nucleotide Nucleotide BLASTP Protein Protein BLASTX Nucleotide=Protein Protein TBLASTN Protein Nucleotide=>Protein TBLASTX Nucleotide=Protein Nucleotide=>Protein --------------------------------------------------------------------------------- Nucleotide=>Protein: Input file is nucleotide, which is translated into protein Before BLAST is run.
How to run online NCBI BLAST? http://blast.ncbi.nlm.nih.gov/Blast.cgi NM_028016 mouse Nanog Gene XM_575662 rat Nanog Gene What are the homologous genes in human genome? What should I do if I want to find all mouse genes that are homologous to human genes?
NCBI BLAST Guide: http://www.ncbi.nlm.nih.gov/blast/BLAST_guide.pdf
How to run Standalone BLAST? http://www.ncbi.nlm.nih.gov/BLAST/download.shtml blast archives contain utilities that allow you to run searches on your own computer. netblast archives contain a command-line network client that allows you to submit searches to NCBI.
How to run BLAST? $ blastall -p blastn –d database.fasta –I query.fast -o output_file For long time run (it is still running even if you log out): $ nohupblastall -p blastn –d database.fasta –I query.fast -o output_file & The most important options: -p "blastp", "blastn", "blastx", "tblastn", or "tblastx". -d database.fasta -iquery.fasta-e Expectation value (E) [Real] default = 10.0 -o output file - -F Filter query sequence (DUST with blastn, SEG with others) [String] -S DNA strand, 3 is both, 1 is top, 2 is bottom [Integer]. default = 3 -gPerforms gapped alignment (not available with tblastx) -W Specifies the word size, >= 4. -G Specifies the gap opening cost, e.g. –G 3 ---is to reduce penalty to 3 There are more than 40 options to use
The database requires to be formatted before BLAST can be run How do I format the database? formatdb -iprotein_seqs.fasta -p T -o T formatdb -iDNA_seqs.fasta –p F -o T -p Type of file T - protein F - nucleotide [T/F] Optional default = T -o Parse options T - True: Parse SeqId and create indexes. F - False: Do not parse SeqId. Many other options, for example, -a Input file is database in ASN.1 format
How to parse the the results from BLAST? Project 1---part I, which is what you are working?
How blast work?------BLAST is A heuristic Algorithm It is too slow to use dynamic programming (DP) to find the homologous sequences in the Genome. That is the reason that BLAST is developed. BLAST is 50 times faster than DP. Seeding: the neighborhood of a word contains the word itself and all other words in the neighborhood whose score is at least as big as T when compared via the scoring matrix. GLKFA----.GLK, LKF, KFA 2. Extension: once the search space is seeded. Alignment can be generated from the Individual seeds. Evaluation: Once the seeds are extended in both directions to create alignments. The alignments are evaluated to determine if they are statistically significant. These that are significant are termed as HSPs Evaluation is partially done by Needleman-Wunsch and Smith-waterman Algorithms
Global alignment Needleman-Wunsch Algorithm COELACANTH P-ELICAN--
Fill the matrix and find the path with max score: Path COELACANTH -PELICAN—- -PELICAN—-
Smith-waterman algorithm The edges start with 0 rather than increasing gap penalities No score is less than 0 The trace-back start from the highest score in the matrix
It is too slow to use dynamic programming (DP) to find the homologous sequences in the Genome. That is the reason that heuristic BLAST is developed. BLAST is 50 times faster than DP.
Other evaluation criteria: E—Expect: the number of alignments expected by chance M,n ---search space (m*n) -----normalized score K----a minor constant ----the probability of symbol i ----the probability of paired symbol I and j
BLAT When to use it: Look for 95% or greater identity for DNA Look for 80% or greater identity for protein Flexible Output: Tab-delimited text file that describe the alignment, but not including the Sequences 2. Can produce NCBI-BLAST and WU-BLAST comparable output
How to use BLAT options $blat database query [-ooc=11.ooc] output.psl -occ=11.ooc tell the program to load over-occuring 11-mers from external file, which increases the speed by a factor of 40 in many cases -t=database type, which can be dna, prot, dnax, dnax---DNA seauence translated in six frames to protein rnax---RNA sequence translated in six frame to protein -q=Query type, which can be dna, prot, dnax, dnax---DNA seauence translated in six frames to protein rnax---RNA sequence translated in six frame to protein
-tileSize sets the size of match that triggers an alignment -stepSize space between the tiles, default is the tilesize -oneOff=N if N=1, allows one mismatch in the tiles but still triggers the alignment. -minMatch=N Sets the number of tile matches. More options are described at http://genome.ucsc.edu/goldenPath/help/blatSpec.html
Here are some blat settings for common usage scenarios: • 1) Mapping ESTs to the genome within the same species • -ooc=11.ooc • 2) Mapping full length mRNAs to the genome in the same species • -ooc=11.ooc -fine -q=rna • 3) Mapping ESTs to the genome across species • -q=dnax -t=dnax • 4) Mapping mRNA to the genome across species • -q=rnax -t=dnax • 5) Mapping proteins to the genome • -q=prot -t=dnax • 6) Mapping DNA to DNA in the same species • -ooc=11.ooc -fastMap • 7) Mapping DNA from one species to another species • -q=dnax -t=dnax • When mapping DNA from one species to another the • query side of the alignment should be cut up into chunks • of 25kb or less for best performance.
BLAT is similar in many ways to BLAST. The program rapidly scans for relatively short matches (hits), and extends these into high-scoring pairs (HSPs) How it works? Blat uses the index to find regions in the genome likely to be homologous to the query sequence. II. It performs an alignment between homologous regions. III. It stitches together these aligned regions (often exons) into larger alignments (typically genes). IV. Finally, BLAT revisits small internal exons possibly missed at the first stage and adjusts large gap boundaries that have canonical splice sites where feasible.
FASTA (Pearson and Lipman 1988) MegaBLAST (Zhang et al. 2000), Sim4 (Florea et al. 1998) does a fine job of cDNA alignment. The SAM program (Karplus et al. 1998) and PSI-BLAST (Altschul et al. 1997) slowly but surely find remote homologs. Gotoh's many algorithms robustly deal with gaps (Gotoh 1990, 2000). SSAHA (Ning et al. 2001) maps sequence reads to the genome with blazing efficiency.
How FASTA algorithm works? The FASTA algorithm allows for the comparison of a query sequence to a DNA sequence database. The algorithm uses a fast search to initially identify sequences from the database with a high degree of similarity to the query sequence. Then it conducts a second comparison on the selected sequences. While FastA is actually just a fast approximation to the Smith-Waterman algorithm, it is slower and more sensitive than the BLAST algorithm because FASTA tolerates gaps in the aligned sequences. http://www-bimas.cit.nih.gov/fastainfo/fasta_algo http://en.wikipedia.org/wiki/FASTA
Position specific iterative BLAST (PSI-BLAST) Refers to a feature of BLAST 2.0 in which a profile (or position specific scoring matrix, PSSM) is constructed (automatically) from a multiple alignment of the highest scoring hits in an initial BLAST search. The PSSM is generated by calculating position-specific scores for each position in the alignment. Highly conserved positions receive high scores and weakly conserved positions receive scores near zero. The profile is used to perform a second (etc.) BLAST search and the results of each "iteration" used to refine the profile. This iterative searching strategy results in increased sensitivity. The tutorial illustrates the potential for PSI-BLAST searches to identify even weak (subtle) homologies to annotated entries in the database. It demonstrates that PSI-BLAST is an important tool for predicting both biochemical activities and function from sequence relationships. ftp ://ncbi.nlm.nih.gov/blast/executables PSI-BLAST uses the blastp program exclusively. How to run? http://blast.ncbi.nlm.nih.gov/Blast.cgi Tutorial: http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/psi1.html
MEGA-BLAST Mega BLAST uses a greedy algorithm [1] for the nucleotide sequence alignment search. This program is optimized for aligning sequences that differ slightly as a result of sequencing or other similar "errors". When larger word size is used , it is up to 10 times faster than more common sequence similarity programs. Mega BLAST is also able to efficiently handle much longer DNA sequences than the blastn program of traditional BLAST algorithm.
Sequence Search and Alignment by Hashing Algorithm (SSAHA): SSAHA is an algorithm for very fast matching and alignment of DNA sequences. It achieves its fast search speed by encoding sequence information in a perfect hash function. The SSAHA algorithm is used to identify regions of high similarity which are then aligned using a banded Smith-Waterman algorithm http://www.sanger.ac.uk/Software/analysis/SSAHA2/




















