Download

1 / 27
270 likes | 461 Views
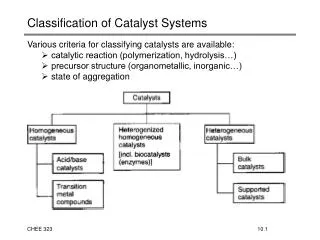
Chaps(14-15) Hetero/Homogeneous Equilibria. • Aqueous Equilibria: Acid-Base Reaction Bronsted_Lowry or proton Transfer Reactions Acid donates H + Base accepts H + AH(aq)+H 2 O(aq) H 3 O + (aq) + A - (aq) K a =exp{- G°/RT} acid base conjugate conjugate

E N D
Chaps(14-15) Hetero/Homogeneous Equilibria • Aqueous Equilibria: Acid-Base Reaction Bronsted_Lowry or proton Transfer Reactions Acid donates H+ Base accepts H+ AH(aq)+H2O(aq)H3O+(aq) + A-(aq) Ka=exp{-G°/RT} acid base conjugate conjugate acid base • pH=-log10[H3O+]: Acidic Solns pH< 7 Basic Solns pH>7 Neutral Soln pH=7 Pure water • Weak Acid/Base Equilibria: Ka< 1 and Kb<1 •Buffered solutions: pH=pKa – log10[HA]0/[A-]0 Final Exam: Monday, June 11th CS 50; 3:00 - 6:00 pm Ch 9, 10, (11-1-11.3, 11.5), (12.1.-.7), 13(Ex .5 & .8), 14(Ex .4 & .8), 15.-15.3 and 18 2 pages of hand written notes, Review Session: Thursday, June 7, 2012, 6:00pm to 7:50pm LAKRETZ 110
Electron Translation and Atomic Vibrations(phonons) Store Energy in Metals By the 3rd Law S 0 as T 0 Standard Entropy with phase transition and P=const T S°(T) =0∫ ncpdT/T + nSfus + nSvap where Svap= Hvap/TB and Sfus= Hfus/TB
S=S2– S1 for Heat Transfer Processes at P=const and V=const Much easier to use S =∫dqrev/T than S=kln For Isothermal Processes: S =(1/T) ∫dqrev=(1/T) qrev qrev=∫dqrev For heat transfer at V=const with no reactions or phase transitions dqrev = ncvdT and S =∫ ncvdT/T = ∫ ncvd(lnT) If cv≠f(T) then S =ncv lnT2/T1 For heat transfer at P=const with no reactions or phase transitions dqrev = ncpdT and S =∫ ncpdT/T = ∫ ncpd(lnT) If cv≠f(T) then S =ncplnT2/T1
So in a Spontaneous Process the heat absorbed by the system from the surroundings is always less than TS The Clausius Inequality S=qrev / T S > qirr / T S ≥ q / T where q is the heat transferred to the system reversibly or irreversibly So for an Isolated system, e.g., the Universe, since q=0 S ≥ 0 for all processes
Consider an Isothermally process @ T with P=const Since quni=0 and Suni = Ssys + Ssur ≥ 0 That is even if quni≠0 Suni≥ quni /T ≥ 0 Suni> 0 for Irreversible Processes(Spontaneous) Suni= 0 for Reversible Processes(equilibrium) Suni< 0 not possible (reverse process is spontaneous) Ssur = qsur /T= – qsys /T = – Hsys /T Ssys– Hsys /T ≥ 0 or [Hsys – TSsys]/T ≤ 0 Define the Gibbs Free energy as G=H - TS @ P and T constant G=[Hsys –TSsys] ≤ 0 or G ≤ 0)
For the Universe Suni> 0 for Irreversible Processes(Spontaneous) Suni= 0 for Reversible Processes(Equilibrium) Suni< 0 not possible(reverse process is spontaneous) For the System G< 0 for Irreversible Processes(Spontaneous) G= 0 for Reversible Processes(Equilibrium) G> 0 not possible(Reverse Process is Spontaneous)
Entropy of Mixing(H=0) System A/B separated by an impermeable membrane: No flow of matter until after the membrane is removed. The surroundings maintain the fixed T and P. Once the membrane is removed A and B will mix spontaneously by diffusion S=S(mixed) – S(unmixed) > 0 G=[H –TS]<0, andH=0 for simple mixing of rare gases for example! So the final state is more disordered than the initial state, which Explains colligative properties like Osmosis G=–TS<0 since S>0 Now cases for H≠0 phase transitions Melting , Boiling etc Thermodynamic Universe
Spontaneous Phase Transitions G(g) G(l) liquid gas G < 0 G < 0 G gas liquid Temperature
Phase Transitions Equilibrium G(g) G(l) G liquid gas Tb Temperature
G=0 for Coexistence Curves. Gas and Liquid phase have the same G above the critical pt. and all 3 phase @ Triple pt . Now Consider H2O(l)H2O(s) H= - Hfus H< 0 S= H/Tm= – Hfus/Tm<0 S= – Sfu< 0 G = -Hfus – T(-Sfu) G = -Hfus + TSfu
Chemical Reactions, the Standard Free energy G° Change can be calculate using Hess’ Law us the standard free energy of formation Gf° which is zero for elements in their standard states Gf° [N2(g)]=0, etc.. G°=H° - TS° For the combustion reaction: CH4(g) + 2O2 (g) CO2(g) + 2H2O(l) G° =? G°={2Gf°[H2O(l)]+Gf°[CO2(g)]} - {Gf°[CH4(g)]+ 2Gf°[O2(g)]} Gf°[O2(g)]=0 And H°={2Hf°[H2O(l)]+Hf°[CO2(g)]} - {Hf°[CH4(g)]+ 2Hf°[O2(g)]} Hf°[O2(g)]=0 However, the entropy is not zero for the elements in their standard states! S°[O2(g)]=S°[O2(g)]=205.3 JK-1≠ 0 S°={2S°[H2O(l)]+S°[CO2(g)]} - {S°[CH4(g)]+ 2S°[O2(g)]}
Assuming again that H° and S° are Independent T If both H° and S° are Negative or positive There is a T*=H°/S° where G° =0 if H° and S° < 0 The rxn is spontaneous T<T* etc. H° and S° > 0 rxn is Spontaneous T>T*. H°> 0 and S°< 0 Rxn is never spontaneous! H°< 0 and S° > 0 the Rxn is always spontaneous!
The Law of Mass Action: aA(g)bB(g) @ Equilibrium K= (PB)b/(PA)a P(atm) aA(aq)bB(aq) @ Equilibrium K=kf/kr= [B]b/ [A]a [molar] Which can be determined from the rate laws of the overall forward and reverse reactions. Can also be determined from thermodynamics K(T)=exp{-G°/RT}
Consider the Reaction dD(g)bB(g) At a pressure PD and PB for products dD(g)bB(g) but H° and G° etc are for P=1 atm need H° at others pressures. This can be determined since enthalpy is a state function! G bB(PB) dD(PD) G2 G1 dD(PD=1 atm) bB(PB=1 atm) G° G= G1 +G° + G 2
Reaction Among Ideal Gases at P≠1 atm, i,e, Not at Standard Pressure conditions G= H – TS G= – TS, since H = 0 S=q/T and q= nRTln(V2/V1) G= –nRTln(V2/V1) Now with P=nRT/V G= –nRTln(P1/P2) G= nRTln(P2/P1) Let P1= Pref G= nRTln(P/Pref ) If Pref=1 atm G= nRTln(P ) P1 P2
dD (PD) bB (PB) dD (PD=1atm) bB (PB=1atm) dD(g) bB(g) G3= bTRln(PB/1) G3= RTln(PB)b G1= dRTln(1/PD) G1= Rln(1/PD)d G2= G°= bGf°[B] - dGf°[D] G= G1 + G° + G3 =G°+RT{ln(1/PD)d + ln(PB)b}= G° +RT{ln(PB)b/(PD)a} G = G° +RT{ln(PB)b/(PD)d} Let Q= (PB)b/PD)d be the reaction Quotient So the Reaction not at Equilibrium G = G° +RT{lnQ}≠0 Fig. 14-4, p. 581
Reaction not at EquilibriumdD(g) bB(g) G = G° +RT{lnQ} Reaction at EquilibriumdD(g) bB(g) G =0= G° +RT{lnQ} and G° = -RTlnK From Kinetics at Equilibrium K= (PBeq)b/PDeq)d Therefore at Equilibrium Q=K and G° = -RTlnK or K(T) = exp{G°/RT}= (PBeq)b/PDeq)a
In GeneraldD(g) bB(g) G = G° +RT{lnQ} and Q= (PB)b/PD)d is the Reaction Quotient G° = -RTlnK then G = -RTlnK+RT{lnQ} = RTln(Q/K) G = RTln(Q/K) If Q<K, Q/K<1 and G <0 dD(g) bB(g) Spontaneous If Q=K, Q/K=1 and G=0 dD(g) bB(g) Equilibrium If Q>K, Q/K>1 and G >0 bB(g) dD(g) Spontaneous
G G>0 G<0 G=0 Gmin Q=K Q<K Q>K
K Small: Reactants favored at Equilibrium Equilibrium to the Left G G<0 G>0 G=0 Gmin Q=K Q>K Q<K Q
K Large: Product Favored Equilibrium to the Right G>0 G<0 G=0 G Gmin Q>K Q<K Q=K Q
For a generic overall Gas Phase Reaction dD + bB eE + fF Q= (PE)e(PF)f/ (PD)d(PB)b And at Equilibrium K= (PEeq)e(Pfeq)f/ (Pdeq)d(PBeq)b Pressure Measured in atm! G° = eGf°[E] + fGf°[F] - bGf°[B] - dGf°[D]
Generalize to Solids, Liquids and Heterogeneous mixed phase reactions. Lets define the activity “a”a=P/Pref SoG= nRTln(P/Pref) goes G= nRTln(a) For none ideal gases ai=i (Pi/Pref)=i Pi Pref= I atm where I = activity the activity coefficient is different For different gases, Ar ≠ Ar Reactions in Solution G= nRTln(a) ai=i ([D]i/[D]ref)= i [D] [D]ref =1 Molar In any case a=1 for pure Solids and Liquids K= (aE)e(aF)f/(aD)d(aB)b
Kinetics Equilibrium [D] [B] dD(g) bB(g) [D]0 [D]eq D K = ([B]eq)b/([D]eq)d [B]eq B
Kinetics Equilibrium [D] [B] dD(g) bB(g) [D]0 [D]eq [D]0 = [D]eq + [D] D K = ([B]eq)b/([D]eq)a K small [B]eq B
Kinetics Equilibrium PD PB dD(g) bB(g) PB=[B]RT (PD)0 (PDeq) (PD)0 = (PD)eq + PA D PD=[D]RT K = (PBeq)b/((PAeq)a K small: Equilibrium towards D: Right (PBeq) B
bB(g) dD(g) The reversed rxn PD or [D] D K’= K-1 = (Pdeq)d/(PBeq)b K’ large, Equilibrium Towards D: left B time