Download

1 / 54
640 likes | 1.31k Views
Lecture 3 Molecular Evolution and Phylogeny. Facts on the molecular basis of life. Every life forms is genome based Genomes evolves There are large numbers of apparently homlogous intra-genomic (paralog) and inter-genomic (ortholog) genes

E N D
Facts on the molecular basis of life • Every life forms is genome based • Genomes evolves • There are large numbers of apparently homlogous intra-genomic (paralog) and inter-genomic (ortholog) genes • Some genes, especially those related to the function of transcription and translation, are common to ALL life forms • The closer two organisms seem to be phylogenetically, the more similar their genomes and corresponding genes are
Central dogma of molecular biology DNA RNA Protein
Basic assumptions of molecular evolution • Closer related organisms have more similar genomes • Highly similar genes are homologs (have the same ancestor) • A universal ancestor exists for all life forms • Molecular difference in homologous genes (or protein sequences) are positively correlated with evolution time • Phylogenetic relation can be expressed by a dendrogram (a “tree”)
The five steps in phylogenetics dancing 1 Sequence data 2 Align Sequences Phylogenetic signal? Patterns—>evolutionary processes? 3 Distances methods Characters based methods Distance calculation (which model?) 4 Choose a method MB ML MP Wheighting? (sites, changes)? Model? Model? Optimality criterion Single tree LS ME NJ Calculate or estimate best fit tree 5 Test phylogenetic reliability Modified from Hillis et al., (1993). Methods in Enzymology 224, 456-487
Why protein phylogenies? • For historical reasons - first sequences... • Most genes encode proteins... • To study protein structure, function and • evolution • Comparing DNA and protein based • phylogenies can be useful • Different genes - e.g. 18S rRNA versus EF-2 protein • Protein encoding gene - codons versus amino acids
Protein were the first molecular sequences to be used for phylogenetic inference Fitch and Margoliash (1967) Construction of phylogenetic trees. Science 155, 279-284.
Most of what follows taken from: Statistical Physics and Biological Information Institute of Theoretical Physics University of California at Santa Barbara 2001 May 7
Understanding trees Root 30 Mya Time 22 Mya 7 Mya same as
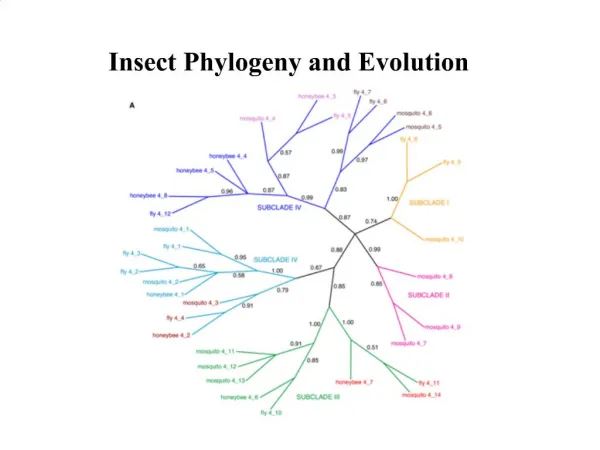
Difference in homologous sequences is a measure of evolution time Part of multiple sequence alignment of Mitochondrial Small Sub-Unit rRNA Full length is ~ 950 11 primate species with mouse as outgroup 靈長目 Change similarity matrix to distance matrix: d = 1 - S
From alignment construct pairwise distance* *Note: Alignment is not the only way to compute distance
Jukes-Cantor (minimal) Model All substitution rates = a all base frequency = 1/4 = 3 Pij(2t) A C
Derivation of Jukes-Cantor formula • Let probability of site being a base at time t be P(t) • After elapse time Dt • mutate to other three bases is –3aDt P(t) • Gain from other bases is aDt (1 - P(t)) • Hence • P(t + Dt) = P(t) –3aDt P(t) + aDt (1 - P(t)) • dP(t)/dt = a - 4a P(t) • Write P(t) = a exp(-bt) +c, solution is b= 4a, c=1/4 • P(t) = a exp(- 4a t) +1/4 • If P(0) = 1, then a = ¾. If P(0) = 0, then a = -1/4 • Finally Psame(t) =1/4 +3/4 exp(- 4a t) Pchange(t) =1/4 - 1/4 exp(- 4a t)
Hasegawa-Kishino-Yano model Has a more general substitution rate Transition A G or C T Transversion A T or C G
Part of Jukes-Cantor distance matrix for primate examples (is much larger; for outgroup) Matrix will be used for clustering methods
Neighbor-Joining Method An Example What is required for the Neighbour joining method? 0. Distance Matrix Distance matrix
1. First Step PAM distance 3.3 (Human - Monkey) is the minimum. So we'll join Human and Monkey to MonHum and we'll calculate the new distances. Mon-Hum Mosquito Spinach Rice Human Monkey
2. Calculation of New Distances After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree. We do this with a simple average of distances: Dist[Spinach, MonHum] = (Dist[Spinach, Monkey] + Dist[Spinach, Human])/2 = (90.8 + 86.3)/2 = 88.55 Mon-Hum Spinach Human Monkey
3. Next Cycle Mos-(Mon-Hum) Mon-Hum Rice Spinach Mosquito Human Monkey
4. Penultimate Cycle Mos-(Mon-Hum) Spin-Rice Mon-Hum Rice Spinach Mosquito Human Monkey
5. Last Joining (Spin-Rice)-(Mos-(Mon-Hum)) Mos-(Mon-Hum) Spin-Rice Mon-Hum Rice Spinach Mosquito Human Monkey
The result:Unrooted Neighbor-Joining Tree Human Spinach Monkey Mosquito Rice
Parsimony criterion Paul Higgs:
Is the best tree much better than others? L: likelihood at nodes
Use Maximum Likelihood to rank alternate trees NJ tree is 2nd best same topology yes yes
Use Parsimony to rank alternate trees different topology ; parsimony differentiates weakly
Clade probability compared from tree methods NJ method is very fast and close to being the best