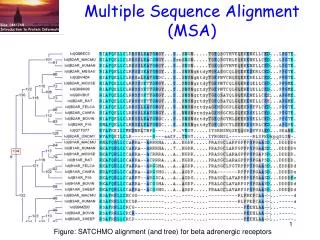
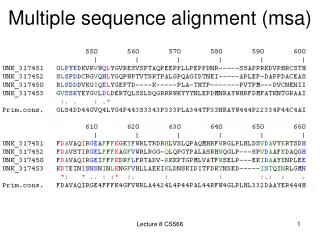
MSA- multiple sequence alignment
MSA- multiple sequence alignment. Aligning many sequences is often preferable to pairwise comparisons. Problem- Computational complexity of multiple alignments grows rapidly with the number of sequences being aligned. .


MSA- multiple sequence alignment
E N D
Presentation Transcript
MSA- multiple sequence alignment • Aligning many sequences is often preferable to pairwise comparisons. • Problem- Computational complexity of multiple alignments grows rapidly with the number of sequences being aligned.
“Even using supercomputers or networks of workstations, multiple sequence alignment is an intractable problem for more than 20 or so sequences of average length and complexity.”
As a result, alignment methods using heuristics have been developed. These methods, (including ClustalW) cannot guarantee an optimal alignment, but can find near-optimal alignments for larger number of sequences.
CLUSTALW • Developed in 1988 • Begins by aligning closely related sequences and then adds increasingly divergent sequences to produce a complete msa.
http://www.ncbi.nlm.nih.gov/ • http://www.ebi.ac.uk/clustalw/
Introduction to Molecular Phylogeny* *Phylogeny- the evolutionary history of a group
Mutations Happen! 3 types possible: • Deleterious • Advantageous • ???
Important Point: • Much of variation that is observed among individuals must have little beneficial or detrimental effect and be essentially selectively neutral. • Deleterious mutations are screened out. Advantageous mutations are rare.
Functional Constraints? • Portions of genes that especially important are said to be under functional constraint and tend to accumulate changes very slowly. • Ex. = histone proteins- practically every amino acid is important. A yeast histone can replace a human histone.
Basis of Molecular Phylogenetics • The evolution of species can be modeled as a bifurcating process- speciation is initiated when two populations become reproductively isolated.
Basis of Molecular Phylogenetics • Once these two populations cease to interbreed, it is inevitable that they diverge due to random mutational processes.
Basis of Molecular Phylogenetics • Over time, this branching process may repeat itself. • A species is said to be related to some other species with which it shares a direct common ancestor.
Basis of Molecular Phylogenetics • The amount of DNA sequence difference between a pair of organisms should indicate how recently those two organisms shared a common ancestor.
Basis of Molecular Phylogenetics • The longer two populations remain reproductively isolated, the more DNA divergence will occur. • The longer two populations remain reproductively isolated, the more protein divergence will occur.
Molecular Phylogeny is relatively new. • Evolution by Natural Selection- Darwin/Wallace 1858 • Molecular Phylogeny 1960s ??
How it started . . .. • In 1959, scientists determined the three-dimensional structures of two proteins that are found in almost every animal: hemoglobin and myoglobin. • During the next two decades, myoglobin and hemoglobin sequences were determined for dozens of mammals, birds, reptiles, amphibians, fish, etc.
What they found . . . • “This tree agreed completely with observations derived from paleontology and anatomy about the common descent of the corresponding organisms.”* • *fromScience and Creationism: A View from the National Academy of Sciences, 2nd Ed., 1999.
Organisms with high degrees of molecular similarity are expected to be more closely related than those that are dissimilar.
Advantages of Molecular Phylogeny • Can be used to decipher relationships between all living things • Relying on anatomy can be misleading- Similar traits can evolve in organisms that are not closely related (i.e. convergent evolution lead to eyes in vertebrates, insects, and molluscs).
Word of Caution Phylogenetic analysis is controversial. There are a wide variety of different methods for analyzing the data, and even the experts often disagree on the best method for analyzing the data.
Why so controversial?? 2 Reasons:
#1 - Molecular vs. Classical • How much weight is given to molecular phylogenetic data, when it contrasts the findings of the traditional taxonomist??
Parking lot “A” at 2:00 Parking lot “A” at 4:00 How many cars changed spaces during this 2 hour interval?
Parking lot “A” at 2:00 Parking lot “A” at 4:00 #2- Molecular Phylogeny requires statistical estimations.
Phylogenetic Data Analysis requires 4 steps • 1) Alignment • 2) Determine the substitution model • 3) Tree Building • 4) Tree Evaluation
STEP 1- Alignment • Molecular phylogenetic analysis is dependent on a good alignment. An evolutionary tree based on an improper alignment is an erroneous tree.
Homology It is critical to phylogenetic analysis that homologous characters be compared across species. Webster’s New Collegiate- Fundamental similarity of structure due to descent from a common ancestral form.
Compare homologous genes and homologous characters: • For DNA and proteins, this means that gaps must be placed correctly in multiple alignments to ensure that the same position is being compared for each species.
Homologous Genes? When could you accidentally compare nonhomologous genes? • Be careful if you comparing genes that are members of a gene family. • Comparing a tubulin-3 from one species with a tubulin-6 from another will not generate accurate results.
What to align? • Phylogenetic trees are generated by comparing DNA or protein. The molecule of choice depends on the question you are attempting to answer.
DNA • contains more evolutionary information than protein : • ATT GCG AAA CAC • * * * * • ATA GCC AAG CTC
Protein (same region analyzed only 1 difference) • Ile-Ala-Lys- His • Ile-Ala-Lys- Leu
DNA • high rate of base substitution makes DNA best for very short term studies, e.g. closely-related species
*Homoplasy • Return of a character to its original state, thus masking intervening mutational events. Every fourth mutation should result in a homoplasy.
Protein • more reliable alignment than DNA: fewer homoplasies than DNA • lower rate of substitution than DNA; better for wide species comparisons
rRNA= ribosomal RNA • Best for very long term evolutionary studies spanning biological kingdoms • Selective processes constraining sequence evolution should be roughly the same across species boundaries
Step 3- Tree Building Tree terminology: Nodes: branching points Branches: lines Topology: branching pattern