Chap.3 Protein Structure & Function
Chap.3 Protein Structure & Function. Topics Hierarchical Structure of Proteins Protein Folding Examples of Protein Function-Ligand-binding Proteins & Enzymes Regulating Protein Function by Protein Degradation Regulating Protein Function by Noncovalent and Covalent Modifications. Goals

Chap.3 Protein Structure & Function
E N D
Presentation Transcript
Chap.3 Protein Structure & Function • Topics • Hierarchical Structure of Proteins • Protein Folding • Examples of Protein Function-Ligand-binding Proteins & Enzymes • Regulating Protein Function by Protein Degradation • Regulating Protein Function by Noncovalent and Covalent Modifications Goals Learn the basic structure and properties of proteins and enzymes, which carry out most of the work in cells (Fig. 3.1).
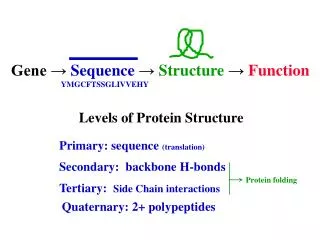
Overview of Protein Structure Hierarchy The four levels of protein structure are illustrated in Fig. 3.2. A detailed discussion of each of these levels is presented in the next few slides. Experiments have shown that the final 3D tertiary structure of a protein ultimately is determined by the primary structure (amino acid sequence). The 3D fold (shape) of the protein determines its function.
Primary Structure The primary structure of a protein refers to its amino acid sequence. Amino acids in peptides (<30 aas) and proteins (typically 200 to 1,000 aas) are joined together by peptide bonds (amide bonds) between the carboxyl and amino groups of adjacent amino acids (Fig. 3.3). The backbone of all proteins consists of a [-N-Ca(R)-C(O)-] repetitive unit. Only the R-group side-chains vary. By convention, protein sequences are written from left-to-right, from the protein’s N- to C-terminus. The average yeast protein contains 466 amino acids. Because the average molecular weight of an amino acid is 113 daltons (Da), the average molecular weight of a yeast protein is 52,728 Da. Note that 1 Da = 1 a.m.u. (1 proton mass). N Ca(R)
Secondary Structure: a Helix Secondary structure refers to short-range, periodic folding elements that are common in proteins. These include the a helix, the b sheet, and turns. In the a helix (Fig. 3.4), the backbone adopts a cylindrical spiral structure in which there are 3.6 aas per turn. The R-groups point out from the helix, and mediate contacts to other structure elements in the folded protein. The helix is stabilized by H-bonds between backbone carbonyl oxygen and amide nitrogen atoms that are oriented parallel to the helix axis. H-bonds occur between residues located in the n and n + 4 positions relative to one another.
Secondary Structure: b Sheets & Turns In b sheets (a.k.a. “pleated sheets”), each b strand adopts an extended conformation (Fig. 3.5). ß strands tend to occur in pairs or multiple copies in b sheets that interact with one another via H-bonds directed perpendicular to the axis of each strand. Carbonyl oxygens and amide nitrogens in the strands form the H-bonds. Strands can orient antiparallel (Fig. 3.5a) or parallel (not shown) to one another in b sheets. R-groups of every other amino acid point up or down relative to the sheet (Fig. 3.5b). Most ß strands in proteins are 5 to 8 aas long. ß Turns consist of 3-4 amino acids that form tight bends (Fig. 3.6). Glycine and proline are common in turns. Longer connecting segments between ß strands are called loops. ß turn
Tertiary Structure Tertiary structure refers to the folded 3D structure of a protein. It is also known as the native structure or active conformation. Tertiary structure mostly is stabilized by noncovalent interactions between secondary structure elements and other internal sequence regions that cannot be classified as a particular type of secondary structure. The folding of proteins is thought to be driven by the need to place the most hydrophobic regions in the interior out of contact with water (Fig. 3.7). The structures of hundreds of proteins have been determined by techniques such as x-ray crystallography and NMR. Different methods of representing structures are shown in Fig. 3.8. Keep in mind that most proteins are somewhat flexible and undergo subtle conformational changes while carrying out their functions.
Secondary Structure Motifs Secondary structure motifs are evolutionarily conserved collections of secondary structure elements which have a defined conformation. They also have a consensus sequence because the aa sequence ultimately determines structure. A given motif can occur in a number of proteins where it carries out the same or similar functions. Some well known examples such as the coiled-coil, EF hand/helix-loop-helix, and zinc-finger motifs are illustrated in Fig. 3.9. These motifs typically mediate protein-protein association, calcium/DNA binding, and DNA or RNA binding, respectively.
Quaternary Structure Multisubunit (multimeric) proteins have another level of structural organization known as quaternary structure. Quaternary structure refers to the number of subunits, their relative positions, and contacts between the individual monomers in a multimeric protein. The quaternary structure of the trimeric hemagglutinin surface protein of influenza virus is shown in Fig. 3.10b. The tertiary structure of a hemagglutinin monomer is shown in Fig. 3.10a.
Modular Domain Structure of Proteins Domains are independently folding and functionally specialized tertiary structure units within a protein. The respective globular and fibrous structural domains of the hemagglutinin monomer (which happen to be individual polypeptide chains) are illustrated above in Fig. 3.10a. Domains (such as the EGF domain) also may be encoded within a single polypeptide chain, as illustrated in Fig. 3.11. Domains still perform their standard functions although fused together in a longer polypeptide (e.g., DNA binding and ATPase domains of a transcription factor). The modular domain structure of many proteins has resulted from the shuffling and splicing together of their coding sequences within longer genes. Epidermal growth factor (EGF) domain
Supramolecular Structure In many cases, multimeric proteins achieve extremely large sizes, e.g., 10s-100s of subunits. Such complexes exhibit the highest level of structural organization known as supramolecular structure. Examples include mRNA transcription preinitiation complexes (Fig. 3.12), ribosomes, proteasomes, and spliceosomes. Typically, supramolecular complexes function as ”macromolecular machines" in reference to the fact that the activities of individual subunits are coordinated in the performance of some overall task (e.g., protein synthesis by the ribosome).
Evolution of Protein Families Through genome sequencing and classical gene cloning approaches, the sequences of an enormous number of proteins have been compiled. Comparison of sequences shows that most proteins belong to larger families that have evolved over time from a common ancestor protein, as illustrated for the globin family of O2 binding proteins (Fig. 3.13). Proteins that have a common ancestor are called homologs. The members of a protein family often show >30% sequence ID, have a common 3D fold, and usually perform closely related functions.
Structure of the Globin Proteins These globular proteins are composed of mostly a helical secondary structure. The similar folds of the globins can be readily seen by comparing the structures of the b chain of hemoglobin, myoglobin, and leghemoglobin (Fig. 3.13). The closely similar structures of mammalian myoglobin and the hemoglobin b subunit might be expected, but the resemblance of the distantly related plant leghemoglobin is striking. Comparison of the sequences of the members of protein families has brought to light the fact that amino acids within a given class exhibit a large degree of functional redundancy. In this regard, the 3 proteins discussed here exhibit less than 20% identity in their sequences, yet have the same structure. Lastly, in hemoglobin 2 different globin chains have combined to form a multisubunit protein.
Overview of Protein Folding Many experiments have shown that proteins can spontaneously fold from an unfolded state to their folded native state. This proves that the amino acid sequence contains enough information to specify tertiary structure. Bonds within the peptide backbone seek out different possible conformations as the final tertiary structure is achieved (Fig. 3.14). Folding tends to occur via successive conformational changes leading to secondary and then tertiary structure elements (Fig. 3.15). The native conformation of a protein typically is its lowest free energy, and therefore, most stable structure. The unfolded (denatured) conformation of a protein can be generated by heating or treatment with certain organic solvents.
Chaperone-assisted Protein Folding The folding of many proteins, particularly large ones, is kinetically slow and is assisted in vivo by folding agents known as chaperones. These proteins are found in all organisms and even in different organelles of eukaryotic cells. Chaperones assist in 1) folding of nascent polypeptides made by translation, and 2) re-folding of proteins denatured by environmental damage, such as heat shock. Molecular chaperones bind to unfolded nascent polypeptide chains as they emerge from the ribosome, and prevent aggregation, misfolding, and degradation (Fig. 3.16a). The hydrolysis of ATP by the chaperone drives conformational changes that prevent aggregation and help drive protein folding. Accessory proteins participate in the process. Eukaryotic molecular chaperones such as Hsp 70 (cytosol & mito matrix) and BiP (ER) are related to the bacterial protein DnaK.
Chaperonins Eukaryotic chaperonins such as the TriC complex are large multimeric complexes related to the bacterial GroEL and GroES proteins. These complexes take up unfolded proteins into an internal chamber for folding (Fig. 3.17). ATP hydrolysis drives folding.
Neurodegenerative Diseases In neurodegenerative diseases such as Alzheimer's disease and transmissible spongiform encephalopathy (mad cow), insoluble misfolded proteins accumulate in the brain in pathological lesions known as plaques, resulting in neurodegeneration (Fig. 3.18). In Alzheimer's disease, the protein known as amyloid precursor protein is cleaved into a peptide product (b-amyloid) that aggregates and precipitates in amyloid filaments. The misfolding of b-amyloid, which involves a transition from a helical to b sheet conformation leads to filament formation. In mad cow disease, prion proteins precipitate causing lesions.
Ligand-binding Proteins The term ligand refers to any molecule that can be bound by a protein. Ligands may be hormones, metabolites, or even other proteins. Ligand binding requires molecular complementarity. The greater the degree of complementarity, the higher the specificity and affinity of the interaction. Affinity is reflected in the Kd for binding. Protein-ligand binding is illustrated here for antibodies (Fig. 3.19a). The complementarity-determining regions (CDRs) of the antibody make highly specific contacts with epitopes in the antigen (Fig. 3.19b). CDR Epitope (a)
Overview of Enzyme Catalysis I Enzymes are proteins (a few are RNAs called ribozymes) that catalyze chemical reactions within living organisms. Enzyme-catalyzed reactions typically are highly specific, and rate enhancements of 106-1012 are common. In an enzyme-catalyzed reaction, the reactant (the substrate) is converted into the product. Like all catalysts, enzymes are not consumed in a reaction. Further, they do not change the ∆G0' or Keq for the reaction, only its rate. Rate enhancement is achieved due to the fact that enzymes are most complementary to the transition state structure formed in the reaction. This results in stabilization of the transition state and lowering of the activation energy barrier (∆G‡) for the reaction (Fig. 3.20).
Overview of Enzyme Catalysis II The transformation of a substrate to the product occurs in the active site of an enzyme. The active site can be subdivided into a catalytic site wherein amino acids that catalyze the reaction reside, and a binding pocket that recognizes a specific feature of the substrate, conferring specificity to the enzyme-substrate interaction. A schematic model for an enzyme catalyzed reaction is shown in Fig. 3.23. The kinetic equation describing the reaction E + S ES E + P. A reaction coordinate diagram showing the binding and catalytic steps of an enzyme catalyzed reaction is shown in Fig. 3.24.
Enzyme Kinetics: Enzyme Concentration The velocity of an enzyme-catalyzed reaction reaches a maximal rate (Vmax) at high concentrations of substrate (Fig. 3.22a). Vmax is achieved when all enzyme molecules have bound the substrate and are engaged in catalysis (saturation). The French mathematicians Michaelis and Menten developed a kinetic equation to explain the behavior of most enzymes. They showed that the maximal rate of an enzyme-catalyzed reaction (Vmax) depends on the concentration of enzyme (Fig. 3.22a) and the rate constant for the rate-limiting step of the reaction. MM equation: Vmax [S] [S] + KM 1.0 x V0 = x x 0.5 x
Enzyme Kinetics: Substrate Affinity Michaelis and Menten also derived a kinetic constant, the Michaelis constant (KM), that is indicative of the affinity of most enzymes for their substrates. The lower the KM the higher the affinity of the enzyme for the substrate (Fig. 3.22b). The KM happens to be the concentration of substrate at which the reaction rate is half-maximal. The concentrations of cellular metabolites usually are set near the KMs of the enzymes that carry out their metabolism. This allows cells to respond to changes in substrate concentration. 1/2 Vmax
Mechanism of Serine Proteases I Proteases are enzymes that cleave peptide bonds in other proteins. The serine proteases, which are important for digestion and blood coagulation, contain reactive serine residues in their catalytic sites. Also present are aspartate and histidine residues that together with serine make up what is called the catalytic triad. The active sites of serine proteases also contain binding pockets that confer specificity by positioning the peptide bond that is to be cleaved next to the reactive serine (Fig. 3.25a, trypsin). The digestive proteases trypsin, chymotrypsin, and elastase select cleavage sites based on the features of their binding pockets (Fig. 3.25b). Specificity Trypsin-basic aas Chymotrypsin-aromatic aas Elastase-small side-chain aas
Mechanism of Serine Proteases II In the serine protease reaction mechanism, an acyl enzyme intermediate is formed transiently after peptide bond cleavage by serine (Fig. 3.26). Subsequently, the acyl group is hydrolyzed off the serine later in the reaction. Both acid-base catalysis (Steps a,c,d,& f) and transition state stabilization (Steps b & e) occur during the reaction. The reaction mechanism is inhibited at low pH due to protonation of His-57 (inset). The pH optimum of serine protease reactions therefore occurs at or slightly above neutrality.
Multifunctional Enzymes Most metabolic pathways occur via multiple enzyme-catalyzed steps. As illustrated in Fig. 3.28, the rates of pathway reactions can be increased if the substrates and products of each step are channeled to the next enzyme in the pathway. Channeling is enhanced in multisubunit enzyme complexes and by attachment of enzymes to scaffolds (Fig. 3.28b), or even by fusion of encoded enzymes into a single polypeptide chain (Fig. 3.28c).
Regulating Protein Function by Degradation The proteolytic degradation (turnover) of proteins is important for regulatory processes, cell renewal, and disposal of denatured and damaged proteins. Lysosomes carry out degradation of endocytosed proteins and retired organelles. Cytoplasmic protein degradation is performed largely by the molecular machine called the proteasome. Proteasomes recognize and degrade ubiquinated proteins (Fig. 3.29). Ubiquitin is a 76-amino-acid protein that after conjugation to the protein, targets it to the proteasome. In ATP-dependent steps, the C-terminus of ubiquitin is covalently attached to a lysine residue in the protein. Polyubiquitination then takes place. The proteasome degrades the protein to peptides, and released ubiquitin molecules are recycling.
Regulating Function by Ligand Binding The binding of a ligand to a protein typically triggers an allosteric ("other shape") conformational change resulting in the modification of its activity. An overview of regulation via allosteric transitions is presented here in the context of the tetrameric O2 binding protein, hemoglobin (Hb). As shown in Fig. 3.30, the O2 binding curve for Hb does not show the simple hyperbolic shape exhibited by proteins that bind a ligand with the same affinity regardless of ligand concentration. Instead, the Hb O2-binding curve is sigmoidal which indicates that the affinity for O2 molecules increases after the first 1 or 2 have bound. In this case, binding displays positive cooperativity. Negative cooperativity is observed with other protein-ligand systems. The reduced O2 binding affinity of Hb at low O2 tensions favors release of O2 to peripheral tissues.
Calmodulin-mediated Switching Many proteins play switching functions in cell signaling. Calcium ion (Ca2+) is a very important messenger in cell signaling. Cells maintain cytoplasmic calcium concentration at about 10-7 M. When calcium concentration rises above this level due to hormone-receptor signaling processes, etc., it binds to a protein known as calmodulin (Kd = 10-6 M) triggering conformational changes that result in its activation. Calmodulin contains 4 helix-loop-helix motifs (EF hands) each of which can bind calcium (Fig. 3.31). Calcium binding causes a major allosteric transition in calmodulin. In its alternate conformation, calmodulin binds to target proteins, changing their activity. Ca2+
GTPase-mediated Switching Proteins belonging to the GTPase superfamily, such as Ras and G proteins, serve as guanine nucleotide-dependent regulatory switches that control of the activity of specific target proteins (Fig. 3.32). When bound to GTP, these proteins adopt an active conformation that modulates target protein function. When bound to GDP, their activity is turned off. The time-frame of activation depends on the intrinsic GTPase activity (the timer function) of these proteins. In addition, GTP and GDP binding (and thus activity) may be regulated by other factors. Examples of such regulation will be covered later. Target protein function
Regulation by Kinase/Phosphatase Switching Protein function also can be regulated by allosteric transitions caused by covalent modification via phosphorylation (Fig. 3.33). Phosphorylation typically occurs on serine, threonine, and tyrosine residues. Enzymes known as kinases carry out phosphorylation. Their activity is opposed by phosphatases, which hydrolyze phosphates off of the modified amino acid. Some proteins are turned on by phosphorylation; others are turned off.