Advanced PCR
Advanced PCR. Dave Palmer, Byotix, Inc. Advanced PCR. PCR of Plant Material Multiplex PCR Modifications to Standard PCR PCR Troubleshooting. Plant DNA Extraction. Rapid 3-Step Extraction Method Freeze TPS 100 mM Tris pH 9.5 1 M KCl 10 mM EDTA Heat 95°C.


Advanced PCR
E N D
Presentation Transcript
Advanced PCR Dave Palmer, Byotix, Inc.
Advanced PCR • PCR of Plant Material • Multiplex PCR • Modifications to Standard PCR • PCR Troubleshooting
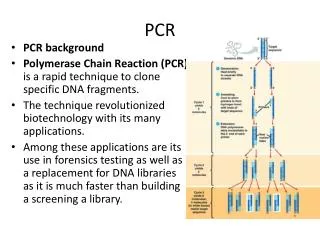
Plant DNA Extraction • Rapid 3-Step Extraction Method • Freeze • TPS • 100 mM Tris pH 9.5 • 1 M KCl • 10 mM EDTA • Heat 95°C D. Thomson and R. Henry, 1995, Single-step protocol for preparation of plant tissue for analysis by PCR, BioTechniques, 19:394-400
How PCR works • Cold Spring Harbor Animation PCR.EXE
Review: The structure of DNA Helix Complementary Base Pairing
Multiplex PCR: What • PCR using several primer pairs SIMULTANEOUSLY • Typically generates a product band for each primer pair
Multiplex PCR: Why • Detect several genes at once • eg. transgenic plant screen • Internal controls • VERY important • Tells you how well the PCR reaction worked • Reduces “false negatives”
Multiplex PCR: How • Same as regular PCR • Care in primer design • Much greater chance of primer-dimers • Annealing temperatures must be close • Much greater chance of artifacts
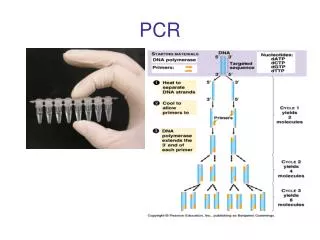
A Typical PCR Reaction • Component ml • Sterile Water 38.0 • 10X PCR Buffer 5.0 • MgCl2 (50mM) 2.5 • dNTP’s (10mM each) 1.0 • PrimerFWD (25 pmol/ul) 1.0 • PrimerREV 1.0 • DNA Polymerase 0.5 • DNA Template 1.0 • Total Volume 50.0
A Typical Multiplex PCR Reaction Component ml Sterile Water 34.0 10X PCR Buffer 5.0 MgCl2 (50mM) 2.5 dNTP’s (10mM each) 1.0 Primer1FWD 1.0 Primer1REV 1.0 Primer2FWD 1.0 Primer2REV 1.0 Primer3FWD 1.0 Primer3REV 1.0 DNA Polymerase 0.5 DNA Template 1.0 Total Volume 50.0
Multiplex PCR: Example • Three primer pairs • Effect, Marker, and Internal Control genes • Control is smallest fragment, also brightest band • Effect is largest fragment, faintest band Effect Gene Marker Gene Control Gene
Multiplex PCR: Example • Three primer pairs Effect Gene Marker Gene Control Gene Which are transgenic?
Multiplex PCR • Problem: • If the control gene product is the BRIGHTEST in a set, then it’s very difficult to tell the difference between weak PCR reactions and nontransgenics. • Solution: • Redesign primers. Effect Gene? Marker Gene? Control Gene
Multiplex PCR: Example • Three primer pairs • Control, marker, and effect genes • Control gene fragment is largest and (almost) faintest • Effect gene is smallest and brightest Control Gene Marker Gene Effect Gene Primers
Multiplex PCR: Example • Three primer pairs Control Gene Marker Gene Effect Gene Primers Which are transgenic?
Other Types of PCR • Different templates • Nested PCR • RT-PCR • Unusual protocols • iPCR • Real-time PCR
PCR Troubleshooting The effect of each component
PCR Reaction Components • Water • Buffer • DNA template • Primers • Nucleotides • Mg++ ions • DNA Polymerase • Extras
Water Purity Contamination Amplification Products PCR Reaction Components
Buffer Must match polymerase Typically contain KCl and Tris Can vary over a slight range: Not much difference in range from 0.8 X to 2.0 X Primer efficiency reduced outside this range PCR Reaction Components http://info.med.yale.edu/genetics/ward/tavi/p06.html
DNA template Amount of DNA present Less DNA means more cycles Complexity of DNA Eg. plasmid vs. whole genome Purity Interfering factors, eg. enzymes, salts Degradation PCR more forgiving of degraded DNA Contamination Amplification products Presence of “poisons” Eg. EDTA which scavenges Mg++ PCR Reaction Components
Primers Age Number of freeze-thaws Contamination Amount Can vary over a wide range (50X) 100-500 nM typical Too low: low amplification Too high: low amplification PCR Reaction Components http://info.med.yale.edu/genetics/ward/tavi/p05.html
Nucleotides 20-400 uM works well Too much: can lead to mispriming and errors Too much: can scavenge Mg++ Too low: faint products Age Number of freeze-thaws Just 3-5 cycles is enough to make PCRs not work well Dilute in buffer (eg. 10mM Tris pH 8.0 to prevent acid hydrolysis) Contamination PCR Reaction Components http://info.med.yale.edu/genetics/ward/tavi/p13.html
Mg++ ions Mg is an essential cofactor of DNA polymerase Amount can vary 0.5 to 3.5 uM suggested Too low: Taq won’t work Too high: mispriming PCR Reaction Components http://info.med.yale.edu/genetics/ward/tavi/p14.html
PCR Reaction Components • Bottom Line: • All components work over a wide range. • Need to avoid contamination. • Optimization by trial-and-error.
DNA Polymerase Thermostable? Activity declines with time at 95C Matches buffer? Age Contamination Concentration: Typically 0.5 to 1.0 U/rxn PCR Reaction Components http://info.med.yale.edu/genetics/ward/tavi/p12.html
Extras Proprietary or added by user Glycerol, DMSO Stabilize Taq, decrease secondary structure May help or hurt, depending on primers Typically already in the Taq stock BSA Frequently helps, doesn’t hurt Betaine Useful for GC-rich templates PCR Reaction Components http://info.med.yale.edu/genetics/ward/tavi/p16.html http://taxonomy.zoology.gla.ac.uk/~rcruicks/additives.html
Denaturation Temp Annealing Temp Extension Temp Time Number of Cycles Reaction Volume “Odd” Protocols PCR Cycling Parameters
Denaturation Step Must balance DNA denaturation with Taq damage 95C for 30 - 60s typically is enough to denature DNA Taq loses activity at high temps: Half-life at 95C: 40 min Half-life at 97.5C: 5 min PCR Cycling Parameters http://info.med.yale.edu/genetics/ward/tavi/p08.html
Annealing Step Most critical step Calculate based on Tm Often does not give expected results Trial-and-Error Almost always must be done anyway Too hot: no products Too cool: non-specific products Gradient thermocyclers very useful Typically only 20s needed for primers to anneal PCR Cycling Parameters http://info.med.yale.edu/genetics/ward/tavi/p08.html
Extension Step Temperature typically 72C Reaction will also work well at 65C or other temps Time (in minutes) roughly equal to size of the largest product in kb Polymerase runs at 60bp/s under optimum conditions Final “long” extension step unnecessary PCR Cycling Parameters http://info.med.yale.edu/genetics/ward/tavi/p08.html http://info.med.yale.edu/genetics/ward/tavi/p10.html
Number of Cycles Number of source molecules: >100,000: 25-30 >10,000: 30-35 >1,000: 35-40 <50: 20-30 fb. nested PCR Do not run more than 40 Virtually no gain Extremely high chance of non-specific products Best optimized by trial-and-error PCR Cycling Parameters http://info.med.yale.edu/genetics/ward/tavi/p08.html
Reaction Volume Doesn’t affect PCR results as long as volume is within limits. Heated lid important. 5ul, 20ul, 100ul all work. Slightly higher yield with lower volumes. PCR Cycling Parameters http://info.med.yale.edu/genetics/ward/tavi/p03.html
“Odd” Protocols Hot-Start PCR Taq is added last Touchdown PCR Annealing temp is progressively reduced PCR Cycling Parameters
Adventures in PCR There’s a fly in my primer! The protocol that never worked.
“There’s A Fly In My Primer!” • This happened when we were first developing PCR methods. • Transgene-specific primers not available for testing. • Needed primers to test DNA extraction protocol, multiplexing. • Synthesized “NS”-series primers • Pastrik, 2000 • A “plant-specific primer set” suitable for potato amplification.
“There’s A Fly In My Primer!” • First Experiment: • DNA extraction and multiplexing • Worked great! • Second Experiment • To confirm first results • Great, but “ghost” bands in controls. No cause for alarm. • Implemented contamination controls.
“There’s A Fly In My Primer!” • Third Experiment • To determine optimum number of cycles • Contamination in controls, blamed on DNA on kimwipes. • Contamination perfectly matched NS band. • Four Experiment • Clear NS bands in control lanes! • Controls only H2O samples. • “What the heck is going on?!”
“There’s A Fly In My Primer!” • Fifth Experiment • Designed to determine where in DNA extraction process the contamination was getting in • Get leaf > Freeze leaf > Add TPS > Cook extract > Spin extract > Add DNA to PCR Tube • Contamination was all across the board, including in UNOPENED PCR TUBES.
“There’s A Fly In My Primer!” • Other Key Observations • Another primer pair (PHY) failed to work during this period of time. • One morning the freezer door was slightly open; didn’t shut properly. • Primers were diluted in H2O (no preservative such as EDTA) • NS primer pair binds to rDNA genes, common across many species (not just potato).
“There’s A Fly In My Primer!” • SO... What Happened? • One or more of the reagent tubes became contaminated. Likely primers. • Contaminating organism was able to multiply a few cycles while the freezer door was open. • Suspect contaminant was an oomycete fungus: has similar “NS” gene. • Extraordinary sensitivity of PCR led to detection of the NS gene in the contaminating reagent!
“There’s A Fly In My Primer!” • Moral of the Story: • PCR is VERY sensitive! • Contamination with template DNA can come from the most unlikely source!
The Protocol That Never Worked • Early PCR method development • We were setting up our PCR lab • Method for DNA extraction and PCR supplied by lab in Holland • Hey, it’s always best to use a protocol that works for someone else, right? • Tried method: • Total failure. No bands at all.
The Protocol That Never Worked • Read about PCR troubleshooting. • Changed Conditions • More cycles. Less cycles. • Hotter. Cooler. • Known good primers. • More Mg++. • New nucleotides. • New polymerase. • All failed.
The Protocol That Never Worked • Needed “Known Good” primers and DNA to properly test the PCR: • Went down the road to USDA-Albany to borrow some known good template DNA. • Had primers known to work on potato DNA synthesized. • IT WORKED! • So That Was A Hint: • Must be something to do with DNA extraction • Either not enough DNA, or some other problem...
The Protocol That Never Worked • Immediately requested original paper that the DNA extraction protocol was based on. • Learned a LOT from reading original paper! • Authors examined effects of all sorts of conditions: • Freezing, temp, salt, EDTA, time, etc. • Varying any condition by up to 50% didn’t substantially affect results. • Therefore we couldn’t have screwed up the extraction. • Comment: “Addition of EDTA requires a compensatory increase in the concentration of MgCl2.”
The Protocol That Never Worked • Quick Calculations: • TPS buffer is 10 mM EDTA • We added 1.0 ul of extract, therefore 10 nmol EDTA. • The protocol called for 1.0 ul of 50 mM MgCl2, therefore 50 nmol Mg++. • One EDTA molecule can bind two Mg++ ions. • Therefore, the TPS buffer was immediately scavenging almost half of the Mg++ we were adding!!!!
The Protocol That Never Worked • Quick Experiment: • Run a Mg++ gradient. • No amplification with 1.0, 1.5 ul MgCl2 • Good amplification with 2.0 and 2.5 ul! • The problem was all in the magnesium • Actually, tried this earlier, but just happened not to try enough the first time... • Never did determine why the Holland lab didn’t have problems.
The Protocol That Never Worked • Moral of the Story: • Just because a protocol works for someone else is NO GUARANTEE it will work for you! • Don’t expect things to work out the first time out. • Consider the troubleshooting to be a “learning process”; you’ll learn SO much MORE if things go wrong and you have to figure out why! • It’s ALWAYS a good idea to go to the original source of the method! • In this case, it was the original scientific paper which had the key to the answer.
A Typical PCR Reaction Sterile Water 38.0 ul 10X PCR Buffer 5.0 ul MgCl2 (50mM) 2.5 ul dNTP’s (10mM each) 1.0 ul PrimerFWD (25 pmol/ul) 1.0 ul PrimerREV 1.0 ul DNA Polymerase 0.5 ul DNA Template 1.0 ul Total Volume 50.0 ul