PCR
PCR. Prof. Vincenzo Nigro Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli. Telethon Institute of Genetics and Medicine (TIGEM). 1978. Prima diagnosi molecolare sul DNA. *Primo ormone umano ricombinante (insulina)


PCR
E N D
Presentation Transcript
PCR Prof. Vincenzo Nigro Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine (TIGEM)
1978 Prima diagnosi molecolare sul DNA *Primo ormone umano ricombinante (insulina) *Premio Nobel agli scopritori degli enzimi di restrizione *Identificato l’AIDS *Prodotto il depakene (epilessia) *Prima bambina in provetta (Luise) *Appare sul mercato l’Apple Computer ed i floppy disk
Com’era l’analisi genetica prima della PCR? • Il Southern blotting (1975) permetteva un’analisi approssimativa dei geni (RFLPs, inserzioni & delezioni) • Il sequenziamento del DNA (1978) richiedeva che I geni venissero prima clonati in appositi vettori (plasmidi o fago l) • La costruzione di genoteche e lo screening potevano richiedere molti mesi e le genoteche dovevano essere preparate per ciascun individuo analizzato
L’invenzione della PCR • Ideata da Kary Mullis nel 1983 • La prima pubblicazione è apparsa nel 1985 • Premio Nobel per la chimica nel 1995
1985 Descritta la reazione di PCR Parte il progetto genoma umano Costo giornale £ 650 Tazzina Caffè £.400
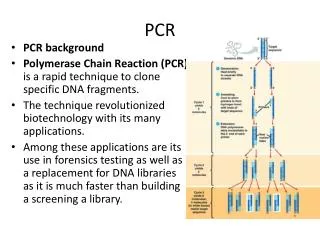
DNA PCR Molte molecole (singola molecola) amplificazione Polymerase Chain Reaction - PCR La PCR rappresenta la seconda rivoluzione nelle tecniche di manipolazione del DNA E’ essenzialmente una tecnica di amplificazione del DNA
Polymerase Chain Reaction • Metodo per l’amplificazione esponenziale di sequenze di DNA • Ingredienti di base • Stampo di DNA o RNA • 2 primers complementari a differenti regioni dello stampo • DNA polimerasitermostabile • 4 nucleotidi • il buffer appropriato
Schema della PCR Estrazione del DNA o RNA dal campione DNA Reverse transcription RNA Amplificazione DNA Rivelazione
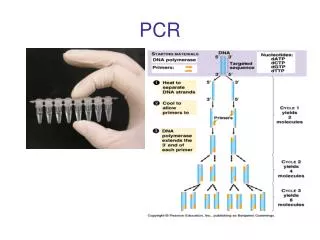
COMPONENTE VOLUME Concentrazione finale 10 X PCR Buffer 5l 1X 10 X dNTPs (2mM) 5l 200M Forward primer (5 pmols/l) 5l 0.5M Reverse primer (5 pmols/l) 5l 0.5M (25pmols/50l) DNA genomico stampo 2l 1g polimerasi termostabile (5U/l) 0.2l 1 unità H2O (a 50l di volume finale) 27.8l TIPICA MISCELA DI REAZIONE 25 o 50l in una provetta micro Eppendorf (0.2 o 0.5 ml)
I cicli della PCR • 30–35 cicli ciascuno comprendente: • denaturazione (95°C), 10-40 sec • annealing (50–65°C), 30-120 sec • polimerizzazione(68-72°C),il tempo dipende dalla lunghezza del frammento
“Tutti I blocchi sono uguali ma alcuni sono più uguali di altri!” Taken from -http://info.med.yale.edu/genetics/ward/tavi/PCR.html
5’ -T A C G • Synthesis by DNA polymerase -A T G C A T G C A T G C * * 3’ T A C G T A Uno specifico DNA a singola elica (primer o innesco) ibrida con l’elica che deve essere copiata
Come funziona la PCR • Come fa la polimerasi a sapere quando fermarsi una volta che ha raggiunto l’altro primer? • PCR animation alla “Dolan DNA Learning Center, CSHL, Cold Spring Harbor”
Quanto è potente la PCR? • La PCR può amplificare fino ad ottenere una quantità utilizzabile di DNA (visibile su gel) in meno di 2 ore • Lo stampo di DNA non necessita di particolari purificazione se il frammento da amplificare è di dimensioni ridotte (fino a 1000bp) • Il prodotto della PCR può essere digerito con enzimi di restrizione, sequenziato o clonato • La PCR può amplificare una singola molecola di DNA (es. uno spermatozoo)
Elettroforesi su gel : Separa le molecule per dimensione separazione orizzontale --> Agarosio = DNA ed RNA separation verticale --> Acrilamide = DNA, proteine ed RNA le molecole più piccole migrano nel gel più velocemente
Elettroforesi su gel: visualizzazione diretta delle molecole separazione orizzontale --> Agarosio = DNA ed RNA
Il DNA colorato con bromuro di etidio emette una fluorescenza di colore rosso-arancio se sottoposto a luce UV
UCSC Genome Browser on Human July 2003 Freeze Screenshot from University of California at Santa Cruz http://genome.ucsc.edu PTEN gene
Quante copie? • Nessun prodotto fino al 3° ciclo • L’accumulazione non è un raddoppiamento completo dopo ciascun ciclo • Dopo 32 cicli ci dovrebbero essere max 1,073,741,764 copie di lunghezza definita (~1109)
How Big A Target? • Amplification products are typically in the size range 100-1500 bp. • Longer targets are amplifiable — >25 kb. • Requires modified reaction buffer, cocktails of polymerases, and longer extension times. • Limited by the integrity of the starting target DNA — > 50 kb.
Designing PCR Primers • Primers should be ~20 bases long. • The G/C content should be 45–55%. • The annealing temperatures should be within 1°C of one another. • The 3´-most base should be a G or C. • The primers must not base pair with each other or with themselves or form hairpins. • Primers must avoid repetitive DNA regions.
Primers That Form Dimers • A primer may form a dimer with itself or with the other primer. 5´-ACCGGTAGCCACGAATTCGT-3´ |||||||||| 3´-TGCTTAAGCACCGATGGCCA-5´ • Primer dimers can be an excellent, but unwanted, substrate for the Taq polymerase.
Primers That Form Hairpins • A primer may be self-complementary and be able to fold into a hairpin: 5´-GTTGACTTGATA ||||| T 3´-GAACTCT • The 3´ end of the primer is base-paired, preventing it annealing to the target DNA.
Optimising the PCR Reaction • Annealing temperature of the primers. • The concentration of Mg2+ in the reaction. • The extension time. • (The denaturing and annealing times.) • (The extension temperature.) • (The amount of template and polymerase— “more is less”.)
Optimising the Annealing Temperature • Primers have a calculated annealing temperature(e.g. 54°C). • Temperature must be confirmed practically. • Temperature steps of 2°C above and below. • Use gradient cycler.
Optimising the Mg2+ Concentration • The fidelity of the PCR depends on [Mg2+]. • Vary [Mg2+] in steps of 0.5 mM. • Sometimes a compromise between yield and specificity.
ADDITIVi? DMSOFormamideGlicerolo QIAGEN – Q Stratagene - Perfect Match
False positive PCR • Contamination • at the time of specimen collection • during transport • in the laboratory from other patient samples or positive control material • negative controls used to detect contamination • Primers are not specific enough • do additional confirmatory tests • PCR for other targets, additional tests to confirm the identity of the product
Fidelity of the Reaction • Taq DNA polymerase lacks the 3´5´ proof-reading activity commonly present in other polymerases. • Taq mis-incorporates 1 base in 104. • A 400 bp target will contain an error in 33% of molecules after 20 cycles. • Error distribution will be random.
Do Errors Matter? • Yes, if you want to clone the amplified DNA — an individual molecule may harbour several mutations. • No, if you want to sequence the amplified DNA or cut it with restriction enzymes. • Use a proof-reading thermo-stable enzyme rather than Taq.
Cloning PCR Products • Products should be ligatable into blunt-ended restriction enzyme site. • Lower than expected efficiency. • Products are not truly blunt-ended. • Taq polymerase adds a single non-templated base (usually A) to the 3´ end: NNNNNNN…NNNNNNNAANNNNNNN…NNNNNNN
Can I PCR Amplify RNA? • Not directly — the DNA polymerase requires a DNA template and will not copy RNA. • mRNA can first be copied into cDNA using reverse transcriptase. • cDNA is a template for PCR — it need not be double-stranded.
Multiplex PCR • PCR reactions can be devised in which several targets are amplified simultaneously — often used in diagnostic applications.