Download
1 / 32
320 likes | 543 Views
AN EXAMPLE FROM MORE ADVANCED BIOINFORMATICS. Gene expression data analysis. VI LEZIONE. Introduzione all'analisi di dati d'espressione genica. Metodi per lo studio dell’espressione genica su larga scala. Profili e matrici d'espressione.

E N D
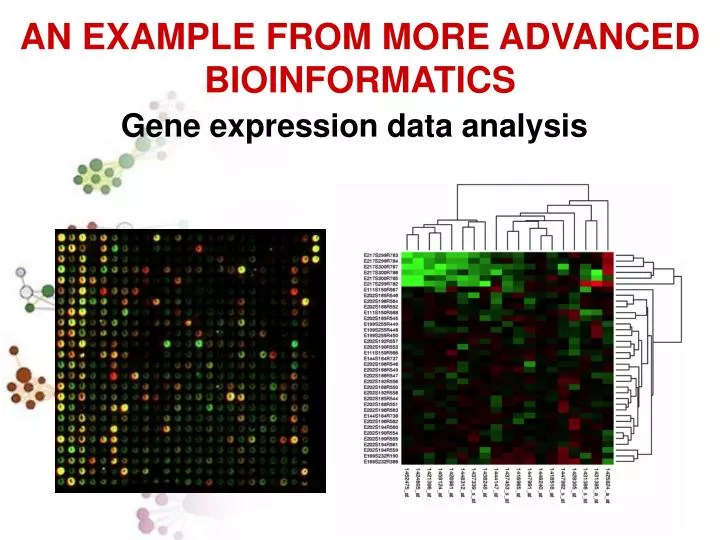
AN EXAMPLE FROM MORE ADVANCED BIOINFORMATICS Gene expression data analysis
VI LEZIONE • Introduzione all'analisi di dati d'espressione genica. • Metodi per lo studio dell’espressione genica su larga scala. • Profili e matrici d'espressione. • Ricerca di geni co-espressi e di geni differenzialmente espressi.
Metodi per lo studio dell’espressione genica su larga scala: • Basati su ibridazione: Microarray/Chip • Basati su conteggio di sequenze: EST sequencing, SAGE, e deep sequencing di librerie di cDNA Deep seq. EST SAGE MICROARRAY CHIP Computational analysis of data by statistical methods
ESPRESSIONE DEL GENOMA UMANO NELLE CELLULE DIFFERENZIATE • Tutte le cellule di un organismo hanno lo stesso corredo genomico • L’espressione genica tessuto specifica determina il fenotipo morfo-funzionale dei tipi cellulari e tissutali • In ogni cellula differenziata ed in ogni particolare momento dello sviluppo e’ attivo solo un sottoinsieme dei geni
REGOLAZIONE DELL’ESPRESSIONE GENICA • Puo’ agire su ciascuno dei livelli che caratterizzano il passare dell’informazione genica dal DNA alle proteine • Negli Eucarioti superiori la regolazione dell’espressione genica si svolge principalmente come controllo della trascrizione • Principali tipi di regolazione: • Controllo epigenetico • Controllo trascrizionale • Controllo post-trascrizionale
“One-gene approach” Il gene di interesse e’ espresso in un tessuto o in un dato momento dello sviluppo ? Quanto e’ attivo dal punto di vista trascrizionale ? Real Time PCR PCR semiquantitativaIbridazione DNA genico o cDNA con RNA totale o poly(A)+RNA (Northern blot)Ibridazione in situ “Large-scale approach” Quali geni sono espressi in un tessuto ed in un dato momento dello sviluppo ? Quanto ciascuno di essi e’ attivo dal punto di vista trascrizionale ? Profilo d’espressione del genoma(TRASCRITTOMA)
METODI PER LO STUDIO SU LARGA SCALA DELL’ESPRESSIONE GENICA BASATI SUL SEQUENZIAMENTO • Sequenziamento sistematico di ESTsda librerie di cDNA • Sequenziamento sistematico con metodi di terza generazione di librerie di cDNA • SAGE (Serial Analysis of Gene Expression)
Deep seq. EST SEQUENCING mRNA of different genes cDNA LIBRARY
EST UniGene Human Release Statistics Total sequences in clusters: 3115711 Total number of clusters sets: 95928 22094sets contain at least one known gene 94710sets contain at least one EST 20876sets contain both genes and ESTs ESTIMATE OF THE LEVEL OF EXPRESSION OF A GIVEN GENE Sample of 12919 ESTs corresponding to 4460 genes/trascripts eg. Rhodopsin: 65 retina ESTs 65 / 12919 = 0.503%
SAGE SAGE Serial Analysis of Gene Expression SAGE è un metodo sperimentale ideato per utilizzare i vantaggi del sequenziamento su larga scala per avere informazioni quantitative di espressione genica (Velculescu et al. 1995, Zhang et al, 1997) Con questa tecnica e’ possibile stimare il livello d’espressione di ciascun gene, attraverso la misura del numero di volte in cui la TAG che lo rappresenta compare in un campione abbastanza grande di TAGs sequenziate a partire dal messaggero del tessuto in analisi Tag to Gene mapping Gene to Tag mapping Consiste nel sequenziamento da messaggeri cellulari di brevi oligonucleotidi, che fungono da etichette di sequenza (TAG)
SAGE • una sequenza di 9 paia di basi permette di identificare 49 (262144) diversi trascritti (una "tag" viene ottenuta da una posizione specifica di ogni trascritto). • le "tag" possono essere unite insieme in serie, a costituire lunghe molecole di DNA, che vengono clonate e sequenziate. • il numero di volte in cui una singola "tag" viene osservata permette di quantificare l'abbondanza del messaggero identificato nella popolazione dei messaggeri e, indirettamente, il livello di espressione del gene corrispondente.
MICROARRAY DUE CANALI Esperimenti di Microarray Permettono l’analisi dell’espressione di migliaia di geni simultaneamente
MICROARRAY DUE CANALI
GeneChip Affymetrix SINGOLO CANALE Ibridizzazione della sonda marcata Scansione del GeneChip con scanner laser
Analisi immagine Normalizzazione Espressione differenziale Clustering Interpretazione biologica
MICROARRAY GeneChip Affymetrix SINGOLO CANALE Analisi dell’immagine • Identificazione della posizione degli spot • Costruzione di un’area locale intorno ad ogni spot • Calcolo dell’intensità di ogni singolo spot • Calcolo del background locale
MICROARRAY GeneChip Affymetrix SINGOLO CANALE Elaborazione dei dati
Deep seq. EST SAGE MICROARRAY CHIP
Matrice dei risultati: righe = geni, colonne = condizioni sperimentali • Quali geni sono differenzialmente espressi ? • Quali e quanti geni sono co-espressi?
Obiettivi dell’analisi saranno… • Identificazione geni differenzialmente espressi • Identificazione pattern di espressione comuni • Identificazione di geni co-espressi con geni di funzione nota
Schema sperimentale “semplice”:Dati d’espressione in colon normale e carcinomaDomanda biologica:Quali geni sono differenzialmente espressi nel confronto ?
GENI DIFFERENZIALMENTE ESPRESSI • Fold Change: un primo criterio puo’ essere quello di identificare i geni la cui espressione nei due gruppi di campioni considerati varia di una certa proporzione (raddoppia, dimezza, …) fold change = 2 • Molti falsi positivi • I geni poco espressi risultano differenzialmente espressi anche3 con variazioni non significative • Selezione basata sui p-values associati a Test T: si applica un test statistico per il confronto delle medie di due campioni a ciascun gene; ogni gene risulta associato ad una probabilità (di essere differenzialmente espresso) • Si esegue uno stesso test statistico molte volte, serve una correzione Uso di metodi basati su permutazioni (SAM) e FDR
Schema sperimentale piu’ complesso:Dati d’espressione in piu’ condizioniDomanda biologica:Posso identificare gruppi di geni espressi in modo simile ?
CLUSTER ANALISI • Il CLUSTERING o analisi cluster o analisi di raggruppamento è un insieme di tecniche di analisi multivariata dei dati volte al raggruppamento di elementi omogenei. • Un insieme di oggetti grande e disomogeo viene classificato in una serie limitata di gruppi omogeneei, ovvero “vicini” in accordo con una specifica misura di distanza.
CLUSTER ANALISI • DUE STEPS: • Misura di similarita’ • Diverse misure • Standardizzazione dei dati • Linking method • criterio per stabilire i gruppi • Metodi gerarchici e non gerarchici
CLUSTER ANALISI I geni sono punti nello spazio: punti vicini nello spazio sono raggruppati insieme • Si parte dalla matrice dei dati X di dimensione nxp e la si trasforma in una matrice nxn di dissimilarità o di distanze tra le n coppie di osservazioni (vettori di p elementi). • Si sceglie poi un algoritmo che definisca le regole su come raggruppare le unità in sottogruppi sulla base delle loro similarità. • Lo scopo e’ di identificare un cero numero di gruppi tali che gli elementi appartenenti ad un gruppo siano – in qualche senso – piu’ simili tra loro che non agli elementi appartenenti ad altri gruppi.
CLUSTER ANALISI Distanza euclidea Correlazione di Pearson
Pearson QT clustering Insieme disomogeneo di 40 geni 6 cluster, gruppi omogenei