Exploring Phylogenetic Data with Splits-Graphs
Phylogenetics Workhop, 16-18 August 2006. Exploring Phylogenetic Data with Splits-Graphs. Barbara Holland. Table 1: North Island road distances. Motivation. When analysing phylogenetic data we usually expect the historical signal to match a tree.


Exploring Phylogenetic Data with Splits-Graphs
E N D
Presentation Transcript
Phylogenetics Workhop, 16-18 August 2006 Exploring Phylogenetic Data with Splits-Graphs Barbara Holland
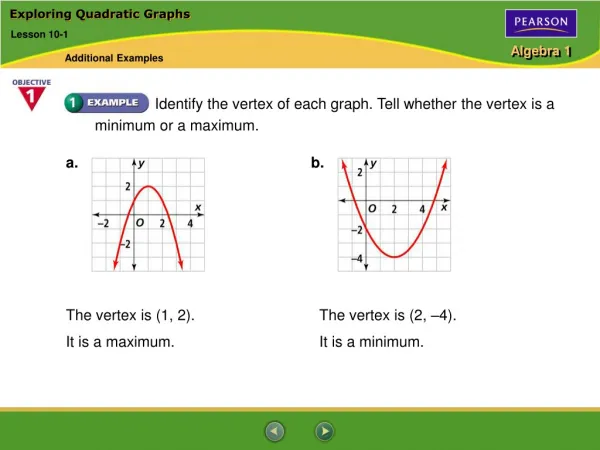
Table 1: North Island road distances Motivation • When analysing phylogenetic data we usually expect the historical signal to match a tree. • So we often use software that specifically outputs a tree. • However, there are many processes that can lead to conflicting signal: • some historical (e.g. hybridisation, recombination); • and some misleading (e.g. long branch attraction, compositional bias, changing patterns of variable sites). • To see if any of these effects are present in our data it is no use using software that can only produce a tree.
Tools • Fortunately, there are a number of tools (some old and some quite recent) that allow conflicting phylogenetic signals to be displayed in a network. • In this talk I will discuss some splits-based methods: • Neighbour Nets, • Consensus Networks and • Spectral Graphs
Splits-based approaches • A split is a bipartition of the taxa (labels) into two sets • A bipartition of one taxa vs. the rest is known as a trivial split • A split corresponds to a branch in a tree • Trees correspond to compatible split systems mouse dog turtle cat, dog, mouse, parrot | turtle parrot cat dog, cat | mouse, turtle, parrot cat, dog, mouse | turtle, parrot
Incompatible splits • Some collections of splits can’t fit on a tree e.g. dog, cat | mouse, turtle, parrot dog, mouse | cat, turtle, parrot turtle, parrot | cat, dog, mouse • But they can fit on a splits-graph mouse dog turtle cat parrot
Split-systems • Different methods produce different varieties of split-systems, e.g. • Tree estimation → Compatible splits • NeighborNet → Circular splits • Split decomposition → Weakly compatible splits • Consensus Networks → k-compatible splits
a b f c e d Circular Splits • Can always be displayed on a planar graph a b c f d e
The same split-system can be represented in different ways b a b a f c f c e d e d abc|def bcd|efa cde|fab
Compatible splits are always circular mouse turtle dog parrot cat owl
Weakly compatible • A split-system is said to be weakly compatible if does not induce on any subset of four taxa all three possible splits. • E.g., the split-system abf|cde ac|bdef ade|bcf Is not weakly compatible as it induces the quartets ab|cd, ac|bd, and ad|bc.
Circular splits are always weakly compatible a √ ab|cd bc|ad √ X ac|bd d b c
k-compatibility • A split-system is said to be k-compatible if there is no subset of k+1 splits that are all pairwise incompatible k=1 k=2 k=3 k=4
Neighbor Net • INPUT: Distance matrix • OUTPUT: A circular split-system, i.e. a split-system that can be displayed as a planar graph. • Runtime: O(n3) • Reference: Bryant, D. and V. Moulton, Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol, 2004. 21(2): p. 255-265.
SELECTION where • Pick a pair of clusters to minimise the standard NJ formula • Choose which node from each cluster are to be made neighbours • Minimise AGGLOMERATION • If a node y has two neighbors x and z, we replace x,y,z with u,v
Consensus Networks • INPUT: (a) a set of leaf-labelled trees, all on the same set of taxa. (b) A threshold t. • OUTPUT: a splits-graph • Runtime: in practice very fast • References:Holland, B., F. Delsuc, and V. Moulton, Visualizing conflicting evolutionary hypotheses in large collections of trees: using consensus networks to study the origins of placentals and hexapods. Syst Biol, 2005. 54(1): p. 66-76.
We have too many trees! • Many phylogenetic methods produce a collection of trees rather than a single best tree. • Monte Carlo Markov Chain (MCMC) • Bootstrapping. • Sometimes trees for different genes produce a collection of trees.
How can we summarize this information? • Large collections of trees can be difficult to interpret. • Consensus tree methods attempt to summarize the information contained within a collection of trees by a single tree. • Information about conflicting hypotheses is necessarily lost.
The problem with consensus trees EXAMPLE: We have 10 trees 5 support the hypothesis ...(gorilla,(human,chimp))... 5 support ...(human,(chimp,gorilla))... None support ...(chimp,(human,gorilla))... In a majority rule consensus tree this would be represented as a polytomy ...(gorilla, human, chimp)... We would lose the information that only 2 of the 3 possible hypothesis have any support in the data. human chimp gorilla human chimp gorilla
Input trees: A D A C E B B C D E D B C C A D D A A B E C E B E Weighted Splits: A,B | C,D,E 2 A,B,C | D,E 2 A,C | B,D,E 1 A,B,D | C,E 1 E D C B A (100%) Strict Consensus tree (>50%) Majority-rule Consensus tree (≥ 33%) Consensus network
Controlling visual complexity • By changing the threshold percentage we can control the worst case complexity of the network. Threshold >50% >33.3% >25% >20%
Why is this so? Example: Given 10 trees and a threshold of 40% the split system will never have 3 mutually incompatible splits. Any split in the split system must be in at least 4 trees. Consider three incompatible splits: By the pigeonhole principle we can see that it is impossible to have 3 mutually incompatible splits
Spectral Graphs • Spectral Graphs exploit the relationship between site patterns in alignments and splits to give a very direct visual representation of a sequence alignment. • Typically an alignment contains many different splits that are not compatible so the resulting splits-graphs tend to be rather complex.
Recoding sites as splits • If a site in an alignment has only 2 states it is easy to see how to recode it as a split. E.g. a …A… b …G… c …G… d …A… ad | bc
Recoding sites as splits • If a site in an alignment has more than 2 states then we need to group states in some way, e.g. purines {A,G} and pyrimidines {C,T}. . a …A… b …G… c …C… d …T… ab | cd
a ab|cd 3 ac|bd 1 ad|bc 1 a|bcd 1 b|cda 1 c|dab 0 d|abc 2 a AGGATTCAG b TGGATCTGG c TAGGTTTAA d TAAGCTCGA b c d Creating the graph • Each split is given a weight proportional to the number of sites that support that split. • Can display all splits or just those splits with weight greater than some threshold.
Example – Rokas et al 2003 • Species phylogeny of 8 yeast based on a concatenation 106 nuclear genes, ~126,000 bps • Found 100% bootstrap support for every edge on the tree • Are all problems in phylogeny solvable with enough data?
NeighborNet of uncorrected distances S. kluyveri S. bayanus S. kudriavzevii C. albicans S. mikatae S. paradoxus S. cerevisiae S. castellii
Consensus Networks of gene trees 106 gene trees from Rokas et al. 2003 Parsimony trees Maximum Likelihood trees S_cerevisiae S_cerevisiae S_paradoxus S_kudriavzevii S_paradoxus S_kudriavzevii S_mikatae S_bayanus S_mikatae S_bayanus S_kluyveri S_kluyveri C_albicans S_castellii C_albicans S_castellii
What have we learned? • Bootstrap support of 100% indicates that sampling error is not a problem, i.e. the result is robust to slight changes in the data. • However, sampling error is not the only source of phylogenetic error and there may still be some strong conflicting signals in the data.
Example 2 – Angiosperm phylogeny • Data taken from Goremykin et al. (MBE, 2004) includes 11 angiosperms • Three gymnosperms for an outgroup • All alignable parts of the chloroplast genome • ~80,000 aligned nucleotide sites for 14 taxa. • Similar to the Rokas example many methods of analysis give high bootstrap support – however, changing the method/model can change the position of the root
NeighborNet Uncorrected distances Grasses Outgroup (gymnosperms)
Neighbornet ML dists (GTR + I + G) Grasses Outgroup (gymnosperms)
Consensus network (parsimony trees) 61 * 1000 = 61,000 bootstrap trees combined Network displays all splits > 6000 trees Support for grasses basal 14,371 / 61,000 Support for Amb +Nym basal 7,203 / 61,000
Maximum Likelihood analysis Each gene fit to GTR + gamma 61 * 100 = 6,100 bootstrap trees combined Network displays all splits > 500 trees Support for Amb +Nym basal 1,277 / 6,100 Support for Nym basal 684 / 6,100 Support for grasses basal 599 / 6,100 Support for Amb basal 574 / 6,100
What have we learned • Long branch attraction is likely to be causing problems for parsimony • Similar to the Rokas data it is probably dangerous to interpret bootstrap scores as measures of accuracy • On the basis of this data there are 4 hypotheses that are still in contention regarding the root of the angiosperm tree.