Download

1 / 6
60 likes | 151 Views
crownether-Host-guest-docked.ppt. Chiral Recognition by NMR Spectroscopy- A Theoretical approach. Illustrating modeling strategies with organic molecules with overtones for Modeling of Bio-molecules An Abstract S.Aravamudhan This material may be available as Internet Resource:

E N D
crownether-Host-guest-docked.ppt Chiral Recognition by NMR Spectroscopy- A Theoretical approach. Illustrating modeling strategies with organic molecules with overtones for Modeling of Bio-molecules An Abstract S.Aravamudhan This material may be available as Internet Resource: Display the web subdirectory: http://www.ugc-inno-nehu.com/CRNMR/ • Some of the linked files in this .ppt file require the Software “ARGUSLAB” be installed in your system. Download the MS Windows installer “setup.exe” file by clicking on the link below: • http://www.ugc-inno-nehu.com/arguslab/ And download the all the contents of this directory into the same and single folder in the resident disc of the P.C. for the hyperlinks in the presentation file to display the appropriate file. Aravamudhan: Chiral Recognition by NMR
While trying to find out the host-guest interactions the thrust has also been to make use of (1) an as much versatile software for the theoretical calculations (2) and a software which is also easily accessible for beginners that would enable them to learn the features of a software for theoretical (Quantum mechanics/ Molecular mechanics) Calculations. Such a report of the facts of investigation would put the beginners on the path for learning about the compromises required in a (macro- ) molecular modeling. Typically the trade off between molecular sizes and available computer memory, together with the necessity to minimize the computational time (which could be a matter of computational costs). The computational chemistry package used for the current work is the online remote computational facility located at http://www.webmo.net . The strong point first of all is the simplicity of access and the student-proof way the web administration manages the Working Demo of this computational chemistry website. The second, but no less important is the fact that this working demo has one of the most advanced features for choosing the computational methods by providing several software engines with each one of them capable of being configured for several properties of molecules to be calculated. Computation, and the way the entire Job parameter can be archived in the resident disc for uploading into the Demo for subsequent calculations at any later time, and the way the results and job parameters can be exported to process and review in off-line mode are not features to which beginners can have access as readily as it is in this working Demo. There is no way as per the current version of the Demo to increase the computational time available per job to more than one minute. This provides an experience in choosing model systems for addressing specific theoretical queries even in the small molecule regime. When the molecule (the host in particular like the 18Crown6Ether) is larger in size most often a MM calculation was preferred for computational time considerations. And, for MM calculations and a few semi empirical QM calculations were possible with the ARGUSLAB more conveniently installed in the PC resident disk. NMR chemical shift calculations were invariably carried out with the online “webmo” remote server computation with ab initio level SCF/STO3G choice for the method and basis set. Aravamudhan: Chiral Recognition by NMR
All G.O. are MM Calculations 18Crown6ether- Flig1-S-F at6 zaxis 18Crown6ether- Flig1-S-F at6 zaxis-UFF-GO hyperlink 18Crown6ether- Flig2-R-Fat6zaxis hyperlink View in Str. Editor View in Str. Editor 18Crown6ether- Flig2-R-Nat6zaxis 18Crown6ether- Flig1-S-N at6 zaxis 18Crown6ether-Flig1-S-Nat6zaxis-UFF-GO hyperlink hyperlink View in Str. Editor View in Str. Editor Aravamudhan: Chiral Recognition by NMR
All G.O. are MM Calculations 18Crown6ether- Flig2-R-Fat6zaxis 18Crown6ether- Flig2-R-Fat6zaxis-UFF-GO hyperlink hyperlink View in Str. Editor View in Str. Editor 18Crown6ether- Flig2-R-Nat6zaxis 18Crown6ether- Flig2-R-Nat6zaxis-UFF-GO hyperlink hyperlink View in Str. Editor View in Str. Editor Aravamudhan: Chiral Recognition by NMR
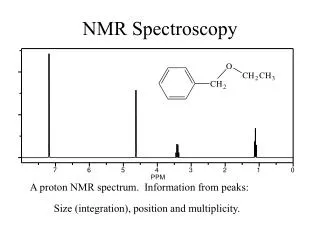
The typical constraint of a time limit of 1 min . per job rendered the G.O. by QM ab initio methods as most inconvenient to opt . However the corresponding geometry ( obtained by the G.O. using any other time saving means) of these molecules could be conveniently put through the one-minute jobs for NMR chemical shift calculations at the “WEBMO”. All the NMR spectra of the different isomers (of the Cs -several lines in spectrum - and of the D3h –)twolines spectrum of the 18Crown6Ether were obtained by ab initio SCF/STO 3G choice with G.I.A .O. for the chemical shift calculation. When it becomes the host (18Crown6ether) –guest (ligand) the NMR calculations exceeded the time limit and hence no calculated spectrum could result for comparison with unbound Host spectrum and the free ligand spectrum (even for the simplest chiral molecule one could envisage as derivative of a methane structure) . Thus even before knowing the actualities of binding criteria which enable the Chiral Discrimination, it became necessary to resort to a modeling strategy, essentially to decrease the size of the molecule and thus in detail know about the theoretical explanations for the possible discrimination. With this situation at the outset, a search for a model. Aravamudhan: Chiral Recognition by NMR