Download
1 / 10
120 likes | 561 Views
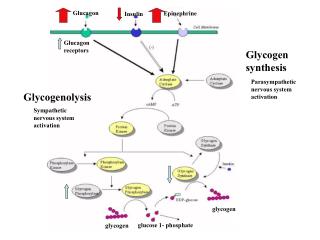
Glucagon. Epinephrine. Insulin. Glycogen synthesis. Glucagon receptors. Parasympathetic nervous system activation. Glycogenolysis. Sympathetic nervous system activation. glycogen. glucose 1- phosphate. glycogen. Etiology: Classification. Inadequate production

E N D
Glucagon Epinephrine Insulin Glycogen synthesis Glucagon receptors Parasympathetic nervous system activation Glycogenolysis Sympathetic nervous system activation glycogen glucose 1- phosphate glycogen
Etiology: Classification • Inadequate production • Increased utilization
Hormonal Response to Hypoglycemia • Suppression of insulin secretion • Glucagon release • Epinephrine and cortisol levels increase • Growth hormone levels increase • Results in suppression of glycogenolysis, activates gluconeogenesis, promotes lipolysis, and stimulates ketogenesis
Etiology: inadequate production of glucose • Prematurity, IUGR, Perinatal stress • Glycogen storage disease • GSD type 1: glucose-6-phosphatase deficiency, catalyzes final common step in glycogenolysis and gluconeogenesis • GSD type 3: debrancher deficiency, inability to degrade stored glycogen. Autosomal recessive and manifestations include hepatomegaly, hypoglycemia, skeletal myopathy, cardiomyopathy
Congenital Hyperinsulinism • Sporadic and familial, incidence from 1/50,000-1/2500 • In the presence of hypoglycemia: • inadequately suppressed insulin level and evidence of excessive insulin action for the degree of hypoglycemia: • inappropriately low plasma ketones and free fatty acids, inappropriately large glycemic response to glucagon.
Congenital Hyperinsulinism • To date, mutations in 4 different genes have been identified • Mutations in either of the two subunits of the ß-cell ATP-sensitive potassium channel (KATP) • Mutations in glucokinase (glucose sensor of the ß-cell) • Mutations in glutamate dehydrogenase • At least 50% of cases have no identified genetic cause
Congenital Hyperinsulinism • Recessive hyperinsulinism • Previous referred to as nesidioblastosis • Islet hyperplasia throughout pancreas • Severe hypoglycemia • LGA newborns • Genetic defect: SUR1 or kir6.2 components of the KATP channel. Thus, therapeutic interventions that act via the SUR such as diazoxide are ineffective • Most infants require near-total pancreatectomy
Congenital Hyperinsulinism • Focal Hyperinsulinism • Area of islet hyperplasia is localized to a small 3-5 mm diameter region described as focal adenomatous hyperplasia • Clinical presentation similar to diffuse disease (severe early hypoglycemia) • Results as a localized clonal loss of the maternal 11p15 region and expression of a paternally inherited SUR1 or Kir6.2 mutation. The 11p15 region contains several maternally imprinted tumor suppressor genes. • Can respond to partial pancreatectomy
Treatment of hyperglycemia • Decrease glucose infusion rate! • Only administer insulin if osmotic diuresis is present or glucose levels remain significantly elevated despite a reduction in glucose infusion rate.