Download
1 / 24
240 likes | 363 Views
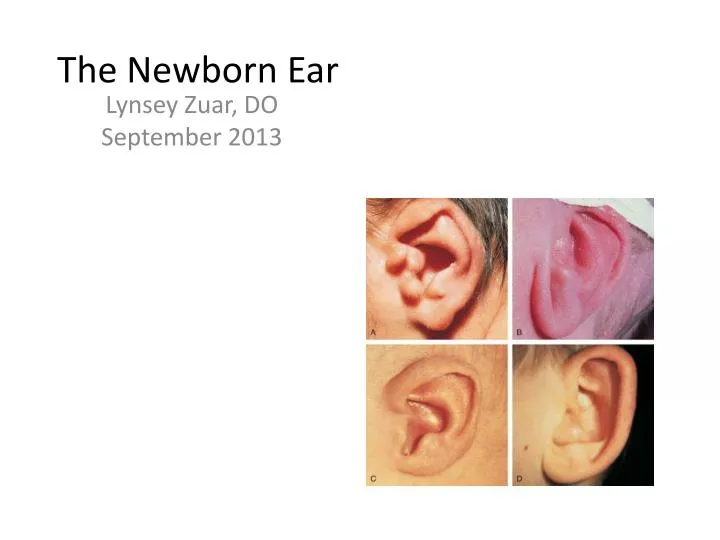
The Newborn Ear. Lynsey Zuar , DO September 2013. Embryological Development of the Outer Ear. External auditory meatus develops from the dorsal portion of the first pharyngeal cleft

E N D
The Newborn Ear LynseyZuar, DO September 2013
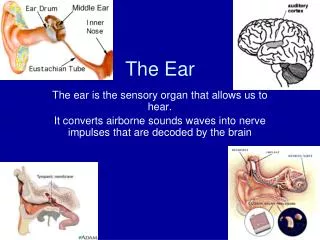
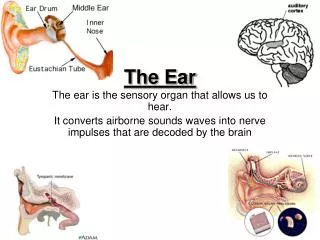
Embryological Development of the Outer Ear • External auditory meatus develops from the dorsal portion of the first pharyngeal cleft • At beginning of the 3rd mo epithelial cells at bottom of meatus form a solid epithelial plate the meatal plug • In 7th mo the plug dissolves and the epithelial lining if the meatus floor helps for the TM • Auricle develops from the 1st & 2nd pharyngeal arches (surrounding the 1st cleft) • Swellings (auricular hillocks) form that eventually fuse to form the auricle can commonly have abnormalities • Initially develop in the lower neck region and migrate up • The development of the ear occurs around the same time as the kidneys (around week 6), and some of the development of the ear and kidneys are dependent on the same genes/chromosomes. • Malformations are seen together usually when a/w other dysmorphic features and syndromes.
The Normal Newborn Ear • Top of ears should be level with outer canthus of eye • Ear cartilage should be formed so that ear holds shape • No preauricular pits, tags, or other deformities should be noted
Preauricular Ear Cysts/Pits/Fissures • Preauricular cysts, pits, and fissures are usually benign congenital malformations of the preauricular soft • Located near the front of the ear – lined with squamous epithelial tissue • Mark the entrance to a sinus tract that may travel under the skin near the ear cartilage • Can produce epithelial-lined subcutaneous cysts or may become infected cellulitis or abscess
Preauricular Skin Tag • Epithelial mounds or pedunculated skin • Arise near the front of the ear around the tragus • No bony, cartilaginous, or cystic components & don’t communicate to the ear canal or middle ear
Cup Ear • Auricle shape that stands away from the head at the superior, posterior, and inferior aspects • In the absence of other abnormalities this finding is only a cosmetic issue • When severe, corrective surgery can be done when the child is older
Microtia Conchal Remnant Type Lobular Type
Microtia aka “Small Ears” • May range from uncomplicated hereditary microtia transmitted as a dominant & harmless trait to severe forms with conductive hearing loss • Non-syndromicmicrotia is AD in a minority of families. • Isolated microtia is uncommon • Abnormal development of the 1st & 2nd brachial arches. • Approximately ½ of babies will have underlying congenital syndrome, so careful assessment for associated abnormalities. Commonly associated with the following: • Isoretinoinembryopathy, FAS, thalidomide embryopathy, maternal DM, Treacher-Collins syndrome, Pierre Robin sequence, or chromosomal syndromes (like Trisomy 18 & 21), congenital infections • Anotia (absence of ear) may be a/w facial paralysis & absence of the tonsil on the abnormal side • Hearing evaluation is mandatory in these infants &referral to a pediatric ENT
Macrotia aka “Big Ears” • Auricle is usually very large but well shaped w/o other ear malformations, & it’s usually b/l & symmetric – may cause psychological disturbance. • Some cases are AD inherited • Commonly associated with the following: • Marfan syndrome, Cerebro-oculo-facial-skeletal syndrome (COFS), Fragile X-syndrome, Variant of Cornelia De Lange type 2 syndrome, Anophthalmia plus syndrome
Lop Ear • A pinna deformity • The superior edge of the helix is folded down • Typically an isolated finding • More of a cosmetic issue • Can be improved with splinting
Stahl's Ear • Pinna has a flat helix at the superior pole, a third crus extending into the helix, and a flattened scaphoidfossa • Can also be a cosmetic concern • Molding or splinting can be initiated by a plastic surgeon – best results in the 1st wk of life • Appearance will stay constant if not corrected • Not a/w with underlying syndromes/conditions
List of Diseases & Syndromes a/w Ear Malformations • Pierre Robin Sequence • Isoretinoinembryopathy • FAS • Thalidomide embryopathy • Maternal DM • Treacher-Collins syndrome • Chromosomal syndromes (like Trisomy 18 & 21) • Congenital infections • CHARGE association • Townes-Brocks syndrome • Branchio-oto-renal syndrome • Nager syndrome • Miller syndrome
When to screen for hearing loss • A 2008 Israeli cross sectional study found an a/w with increased incidence of hearing impairment and isolated preauricular pits and tags (conductive & mixed HI), when compared to healthy infants w/o pits or tags (7x more common) • Evoked Otoacoustic Emission test before discharge of these infants is highly recommended • These infants should also be routinely screened after birth for HI
Evoked Otoacoustic Emission • Otoacoustic emission (OAE) is a sound which is generated from within the inner ear • Evoked otoacoustic emissions (EOAEs) needs an external evoking stimulus • Stimulus Frequency OAEs (SFOAEs): measured during application of a pure-tone, and are detected by the difference between the stimulus waveform and the recorded waveform. • Transient-evoked OAEs (TEOAEs or TrOAEs) are evoked using a broad frequency click or toneburst (brief pure tone) stimulus.
When to screen for kidney dysfunction • A renal u/s should be performed with isolated preauricular pits, cup ears, or any other ear anomaly with 1 or more of the following: • Other malformations/dysmorphic features • Family history of deafness, auricular and/or renal malformations • Maternal history of gestational diabetes. • In absence of above, renal u/s not needed • As mentioned before ear malformations are a/w increased frequency of clinically significant structural renal anomalies compared with the general population. • This is due to observing that auricular malformations often are associated with specific MCA syndromes that have high incidences of renal anomalies.
References • AAP • AAP Journal of Pediatrics • UpToDate • http://newborns.stanford.edu/PhotoGallery/