Download

1 / 1
10 likes | 144 Views
A novel SMAD4 mutation causing severe Juvenile Polyposis syndrome with protein loosing enterophathy and Hereditary Haemorrhagic Telangiectasia. Joel Johansson‡, Christofer Sahin‡, Rebecka Pestoff ¤, Mattias Ekstedt‡, Simone Ignatova†, Pia Forsberg±, Marie Stenmark-Askmalm¤

E N D
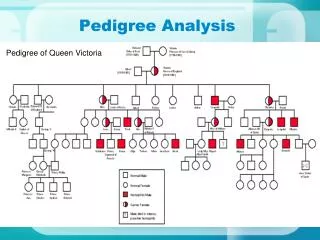
A novel SMAD4 mutation causing severe Juvenile Polyposis syndrome with protein loosing enterophathy and Hereditary Haemorrhagic Telangiectasia. Joel Johansson‡, Christofer Sahin‡, Rebecka Pestoff¤, Mattias Ekstedt‡, Simone Ignatova†, Pia Forsberg±, Marie Stenmark-Askmalm¤ ‡ Gastroenterology and Hepatology unit, ¤Division of Clinical Genetics, † Clinic of Pathology and ±Division of Infectious Diseases ‡ ¤ † ± Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, County Council of Östergötland, Linköping, Sweden Introduction Case Summary Discussion Molecular genetics CONCLUSIONS 1565delC frameshift mutation in C-terminus Protein-protein interaction SMAD4 Figure1: Result from BLAT search with the mutation 1565delC in SMAD4. UCSC Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly • Rare disorders requires complex care-coordination. • These patient’s care is best provided by multidiciplinary team lead by genetics. • Patients with SMAD4 mutation should be followed both for JPS and HHT, and may develope protein loosing enteropathy. Figure 3: Pedigree of the affected family. Only two cases of JPS noted and confirmed with moleculargenetic analysis. Figure 2: SMAD4 trimeric assembly. • Year Age (yrs) Event • 1967 0 Born • 1979 12 Anemia, epistaxis • 1981 14 Rectal bleeding, gastric polyp, colon polyps • 1984 17 Colectomi + IRA. Kulchitskycells = ?Peutz Jeghers • 1986 19 Gastric X-ray = no gastric polyps • 1987-92 20-25 2 ectopic pregnancies • 1992 25 Subileus, 4 polyps of distal ileum. Pregnancy, ↓Fe+, diahorrea • 27 Rectal bleeding, rectal hyperplastic eroded polyp. • 27 Pregnancy: hyperemesis, epistaxis. • Gastroscopy: hamartoma and pyloposis of the ventricle. • 1995 28 Gastrectomy, eosophagojeujunostomi • ? Genetic investigation • Gastric pain = medication with opoids and benzoidazepine • ↓7kg weight, TPN central line = multiple sepsis • 29 Hysterectomy due to sterilization • Science: The SMAD4 gene is located • 1997 30 ↓Fe+, ↓Albumin • Genetic investigation: ?FAP ?JPS • 31 Venferrin, depression • Science: SMAD4 germline mutation associated with JPS • 1999 32 Subileus, ileus, small bowel gangrene = 20 cm resection (no polyps) • 2000 33 Extracts all teeth. Sepsis, pneumonia, drug poisoning=iatrogenic mixed substance abuse • 2005 38 Bilateral pitting oedema, ↓Fe+, ↓Albumin • 2006 39 Dental implants + TPN • Daughter referred to genetics due to anemia, gastric • and colonic polyps • 2008 41 Endoscopy = 2 polyps by IRA (adenomas with low grade dysplasia) • Chronic inflammation of the ventricle • 42 Epigastric pain = gallstone pancreatitis • Ovarial cyst (cystadenofibroma) • Vaginal polyp (fibroepithelial) • ? Genetic diagnosis for patient and daughter • Multidiciplinary care conference • 2012 45 ?JPS • Confirmed novel mutation in SMAD4, c.1565delC • Abdominal ultrasound, 3cm polyp small bowel • ↓Albumin, TPN-resistant • SCIg treatment terminated due to adverse reactions Juvenile polyposis syndrome (JPS) is a rare genetic disorder characterized by juvenile polyps of the gastrointestinal tract; additional extra intestinal manifestations can include telangiectasias. This disorder is most frequently caused by mutations of the SMAD4 or BMRP1A genes. Most of the genes are associated with the BMP/TGF-β signaling pathways. JPS is a clinical diagnosis where one of three criteria must be met: 1. ≥5 juvenile polyps in the large intestine, 2. multiple juvenile polyps throughout the gastrointestinal tract or 3. ≥1 juvenile polyps with juvenile polyps in a first-degree relative. SMAD4 mutations cause JPS complicated with haemorrhagic hereditary telangiectasia (HHT) and in this case protein losing enteropathy (PLE). We here present a case of JPS where misdiagnosis and fragmented care resulted in (preventable) distress for the patient due to doctors delay. This case presents with severe symptoms including protein loosing enteropathy and a novel mutation which has not previously been described in the literature. The patient underwent genetic testing for several genes associated with hereditary gastrointestinal polyps: APC, MUTYH, BMPR1A all gave negative results. Finally, 32 years after presenting with first symptoms, a novel, disease-causing mutation was found in SMAD4, c.1565delC, p.Pro522fs. The mutation is a frameshift mutation in the highly conserved C-terminus and results in a truncated protein 17 amino acids prematurely. ( Figure 1) This alters the binding affinity of the protein, which may affect the homo/heterotrimeric interactions that SMAD4 is involved in. (Figure 2) Studies suggest that 15%-22% of individuals with an SMAD4 mutation are likely to have the combined JPS/HHT syndrome. Loss of function of SMAD4 is the mechanism behind both JPS and HHT, as illustrated by this novel mutation. Studies indicate that any patient with a SMAD4 mutation is at risk for the visceral manifestations of HHT, and of risk for early-onset gastrointestinal cancer. This case illustrates a severe clinical case of polyposis and highlights the importance of multidisciplinary care to reach a correct diagnosis. Clinical care often focuses on symptom relief and not on identifying the underlying cause. Care coordination is therefore of importance, and reduces the risk of stagnant clinical investigations loosing track among multiple treating physicians. When the patient first presented with symptoms genetic diagnosis was not possible, however it could have been performed sooner than it was. Our patient suffered multiple infections, maybe due to general malnutrition and loss of immunoglobulins and smaller proteins. Together with low albumin levels resistant to TPN this implicates protein loosing enteropathy. To our knowledge this is the first reported case of immunodeficiency due to protein loosing enteropathy in JPS. Recurring hemorrhages have also been a running point in the management of this patient, probably due to HHT, linking this novel SMAD4 mutation to both HHT and PLE. We empathize that patients with rare genetic disorders where no definitive treatment is possible or in cases where such disorders are suspected should be managed within multidisciplinary teams with a clinical geneticist or genetic counselor as chairperson. Contact information. Linköping University Hospital: Regional Genetics Clinic, www.lio.se, rebecka.pestoff@lio.se, 2013-08-14