Download

1 / 2
20 likes | 40 Views
Proteinu2013protein interactions play essential roles in various aspects of cellular processes, including metabolic control, signal transduction, gene regulation, and cell communication. Genome-wide proteomics studies provide an increasing list of interacting proteins, but only a small fraction of the potential complexes is amenable to direct experimental analysis. Knowing the structural details of a protein-protein interaction helps us understand the mechanism of such interaction, and thus the function of participating proteins. However, crystal structures of protein complexes are more difficult to get than individual proteins. Furthermore, many protein interactions are transient, which makes studying them by crystallography and NMR even more problematic. Hence, the need for fast and robust computational approaches to reliably predict the structures of proteinu2013protein complexes is growing.

E N D
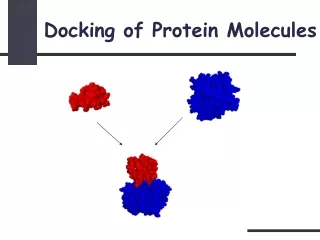
Protein protein docking Protein–Protein Docking INQUIRY Protein–protein interactions play essential roles in various aspects of cellular processes, including metabolic control, signal transduction, gene regulation, and cell communication. Genome-wide proteomics studies provide an increasing list of interacting proteins, but only a small fraction of the potential complexes is amenable to direct experimental analysis. Knowing the structural details of a protein-protein interaction helps us understand the mechanism of such interaction, and thus the function of participating proteins. However, crystal structures of protein complexes are more difficult to get than individual proteins. Furthermore, many protein interactions are transient, which makes studying them by crystallography and NMR even more problematic. Hence, the need for fast and robust computational approaches to reliably predict the structures of protein–protein complexes is growing. Protein–protein docking is the computational prediction of protein complex structure as it would occur in a living organism, given the individually solved component protein structures. Profacgen makes use of state-of-the-art docking software tools to find the relative transformation and conformation of two proteins that result in a stable (energetically favorable) complex. The docking process is composed of two main steps: 1) Generating a set of configurations which reliably includes at least one nearly correct one, and 2) reliably distinguishing nearly correct configurations from the others with a scoring function. Our procedure starts with rigid body global search based on the Fast Fourier Transform (FFT) correlation approach that samples rotational and translational space of one protein while fixing the other. If binding sites are known, this information can be used to reduce the search space. The generated conformations are then scored and ranked based on shape-complementarity, electrostatic and desolvation contributions. Results are clustered to find highly populated low-energy conformations. The representative complexed structure(s) can be further refined by energy minimization. Protein–protein docking process Protein–protein docking process Profacgen employs computational docking techniques to search all possible binding modes in the translational and rotational space between the two proteins and evaluates each pose using an energy-based scoring function. It is
an important means for understanding the physicochemical forces that underlie macromolecular interactions and a valuable tool for modeling protein complex structures at the atomic level. Furthermore, the precise understanding of protein-protein interactions for disease-implicated targets is ever more critical for the rational design of biologic-based therapies. Features Sampling using Fast Fourier Transform (FFT) based methods Scoring function considering shape complementarity, electrostatics, desolvation Filtering output predictions based on blocking residues, binding site residues, pairwise distance restraints Clustering to find highly populated clusters of low-energy conformations High resolution flexible docking for complex structure refinement Identification of hot-spot residues in protein-protein interactions Support modified amino acids in proteins Prediction of the quaternary structure of homo-oligomer Membrane protein docking We provide the service in a customizable fashion to suit our customers’ specific research goals. Please do not hesitate to contact us for more details about our protein–protein docking service.