Download

1 / 35
350 likes | 450 Views
Learn about a probabilistic approach for protein backbone tracing in electron density maps, utilizing a global consistency model to enhance accuracy and efficiency in determining atom coordinates.

E N D
A Probabilistic Approach to Protein Backbone Tracing in Electron Density Maps Frank DiMaio, Jude Shavlik Computer Sciences Department George Phillips Biochemistry Department University of Wisconsin – Madison USA Presented at the Fourteenth Conference on Intelligent Systems for Molecular Biology (ISMB 2006), Fortaleza, Brazil, August 7, 2006
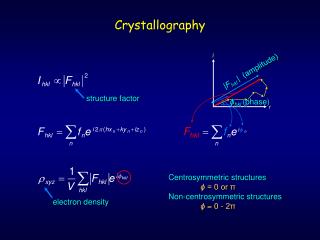
X-ray Crystallography FFT X-ray beam ProteinCrystal CollectionPlate ElectronDensity Map (“3D picture”)
Given: Sequence + Density Map Sequence + Electron Density Map
Our Subtask: Backbone Trace Cα Cα Cα Cα
The Unit Cell • 3D density function ρ(x,y,z) provided over unit cell • Unit cell may contain multiple copies of the protein
The Unit Cell • 3D density function ρ(x,y,z) provided over unit cell • Unit cell may contain multiple copies of the protein
Density Map Resolution 2Å 4Å 3Å ARP/wARP (Perrakis et al. 1997) TEXTAL (Ioerger et al. 1999) Resolve (Terwilliger 2002) Our focus
Overview of ACMI (our method) • Local Match • Algorithm searches for sequence-specific 5-mers centered at each amino acid • Many false positives • Global Consistency • Use probabilistic model to filter false positives • Find most probable backbone trace • Global Consistency • Use probabilistic model to filter false positives • Find most probable backbone trace
5-mer Lookup and Cluster …VKHVLVSPEKIEELIKGY… PDB Cluster 1 Cluster 2 NOTE: can be done in precompute step wt=0.67 wt=0.33
5-mer Search • 6D search (rotation + translation) forrepresentative structures in density map • Compute “similarity” • Computed by Fourier convolution (Cowtan 2001) • Use tuneset to convert similarity score to probability
NEG POS match to tuneset Bayes’ rule score distributions probability distribution over unit cell P(5-mer at ui|Map) search density map scores ti (ui) Convert Scores to Probabilities 5-mer representative
In This Talk… • Where we are now For each amino acid in the protein, we have a probability distribution over the unit cell • Where we are headed Find the backbone layout maximizing
Pairwise Markov Field Models • A type of undirected graphical model • Represent joint probabilities as product ofvertexand edge potentials • Similar to (but more general than) Bayesian networks y u1 u2 u3
Protein Backbone Model • Each vertexis an amino acid • Each label is location + orientation • Evidence y is the electron density map • Each vertex (or observational) potentialcomes from the 5-mer matching ALA GLY LYS LEU
Protein Backbone Model ALA GLY LYS LEU • Two types of edge (or structural) potentials • Adjacency constraints ensure adjacent amino acids are ~3.8Å apart and in the proper orientation
Protein Backbone Model ALA GLY LYS LEU • Two types of structural (edge) potentials • Adjacency constraints ensure adjacent amino acids are ~3.8Å apart and in the proper orientation • Occupancy constraints ensure nonadjacent amino acids do not occupy same 3D space
Backbone Model Potential Constraints between adjacent amino acids: = x
Backbone Model Potential Constraints between nonadjacent amino acids:
Backbone Model Potential Observational (“amino-acid-finder”) probabilities
Probabilistic Inference • Want to find backbone layout that maximizes • Exact methods are intractable • Use belief propagation (BP) to approximate marginal distributions
Belief Propagation (BP) • Iterative, message-passing method (Pearl 1988) • A message, , from amino acid i toamino acid j indicates where i expects to find j • An approximation to the marginal (or belief),is given as the product of incoming messages
Belief Propagation Example ALA GLY
Technical Challenges • Representation of potentials • Store Fourier coefficients in Cartesian space • At each location x, store a single orientation r • Speeding up O(N2X2) naïve implementation • X = the unit cell size (# Fourier coefficients) • N = the number of residues in the protein
Speeding Up O(N2X2) Implementation • O(X2) computation for each occupancy message • Each message must integrate over the unit cell • O(X log X) as multiplication in Fourier space • O(N2) messages computed & stored • Approx N-3 occupancy messages with a single message • O(N) messages using a message product accumulator • Improved implementation O(NX log X)
1XMT at 3Å Resolution prob(AA at location) HIGH 0.82 0.17 1.12Å RMSd 100% coverage LOW
1VMO at 4Å Resolution prob(AA at location) HIGH 0.25 0.02 3.63Å RMSd 72% coverage LOW
1YDH at 3.5Å Resolution prob(AA at location) HIGH 0.27 0.02 1.47Å RMSd 90% coverage LOW
Experiments • Tested ACMI against other map interpretation algorithms: TEXTAL and Resolve • Used ten model-phased maps • Smoothly diminished reflection intensitiesyielding 2.5, 3.0, 3.5, 4.0 Å resolution maps
RMS Deviation ACMI ACMI Textal Resolve Cα RMS Deviation Density Map Resolution
Model Completeness % chain traced % residues identified ACMI ACMI Textal Resolve Density Map Resolution
Per-protein RMS Deviation TEXTAL RMS Error Resolve RMS Error ACMI RMS Error
Conclusions • ACMI effectively combines weakly-matching templates to construct a full model • Produces an accurate trace even with poor-quality density map data • Reduces computational complexity from O(N2X2) to O(NX log X) • Inference possible for even large unit cells
Future Work • Improve “amino-acid-finding” algorithm • Incorporate sidechain placement / refinement • Manage missing data • Disordered regions • Only exterior visible (e.g., in CryoEM)
Acknowledgements • Ameet Soni • Craig Bingman • NLM grants 1R01 LM008796 and 1T15 LM007359