Download

1 / 24
240 likes | 405 Views
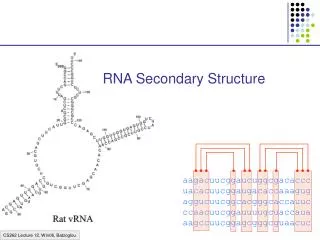
Phylogenetic Reconstruction based on RNA Secondary Structural Alignment. Benny Chor, Tel-Aviv Univ. Joint work with Moran Cabili, Assaf Meirovich, and Metsada Pasmanik-Chor. Phylogenetic Trees Based on What ? Morphology (1800 - ) Single gene sequence (DNA or AA) (1960 - ).

E N D
Phylogenetic Reconstruction based on RNA Secondary Structural Alignment Benny Chor, Tel-Aviv Univ. Joint work with Moran Cabili, Assaf Meirovich, and Metsada Pasmanik-Chor
Phylogenetic TreesBased on What ? • Morphology • (1800 - ) • Single gene sequence (DNA or AA) • (1960 - )
Phylogenetic TreesBased on What ? • Whole genomes • (2002 - )
More Sources to Base Phylogeny On? A Proposed, Metric Induced Approach • 1. Finda reliable metric between pairs of objects. • Design / choose / modify a good algorithm for determining metric (pairwise distances). • Compute distance matrix. • 4. Construct a Neighbor Joiningtree from the distance matrix. • 5. As a sanity check, compare resulting tree to • “standard & accepted” ones. NJ
Was already applied (fairly successfully), e.g. for constructing phylogenies based on whole genomes/proteomes (Burstein et al., 2005), and others, based on metabolic networks (Tuller et al., 2006). • Metric Induced Approach NJ Of course distances that are appropriate to each domain must be applied (or especially designed).
Our Question • Can phylogenetic reconstruction be based on RNA secondary structures ?
Our tree, based on secondary structs. of 16s rRNA from 91 species Answer: Yes, And Even Quite Well Archaea Eukarya Bacteria
Metric Induced Approach: Specifics • Find an efficient alignment algorithm • (similarity based) pair-wise RNA secondary • structures. • 2. Transform similarity to distance. • Use RNA databases to get the RNA molecules • and structures. Apply the algorithm to compute • the distance for each pair of molecules. • 4. Run NJ to produce trees.
The Alignment Algorithm Chosen • We chose to use RSmatch: A sophisticated dynamic programming algorithm, based on the “dot bracket” representation of the secondary structure. • J. Liu , J.T. Wang , J. Hu , B. Tian. BMC Bioinformatics 2005 , 6:89. • RSmatch sorts each dot and bracket to components, and then compares components according to their order in the secondary structure. • RSmatch employs both sequences and structures. • Complexity: O(nm), where n and m are the lengths of the two RNA molecules that are compared. TAATTATCGGAAGCAGTGCCTTCCATAATTA ( ( ( ( ( ( ( . ( ( ( ( ( . . . . . . ) ) ) ) ) ) ) ) ) ) ) )
From Similarity to Distance In transforming the scoring matrix from similarity to distance, we tried to preserve the ratios between mismatches values, and of course lower similarity should imply higher distance. Distance metric requirements: Symmetry, Δ inequality, non negativity, self distance=0
Actual Distance Matrices: Higher Mismatch Penalties at “Dots” - Gap cost : 3 per nucleotide involved. - Δ inequality : mismatch < 2* gap cost
DBs constructed with manual intervention • RNaseP DB: • http://www.mbio.ncsu.edu/RNaseP/ • Sequences length: ~300 - 400 (+/-) nucleotides RNaseP function: Cleaves off an extra, or precursor, sequence of RNA on tRNA molecules. • DBs of Reliable Secondary Struc. • 16S rRNA: • Comparative RNA Web Site: http://www.rna.icmb.utexas.edu/ • Sequences length: ~1,500 (+/-) nucleotides 16S function: In charge of tRNA binding and formation of peptide bonds during translation.
RNaseP Tree, 51 Species • Secondary structure based tree • Good partition to 3 • kingdoms. • Bacteria • (characterized by • Bxy) also look good.
Eukarya Bacteria • RNaseP 51 Species • Sequence based tree Archaea Eukaryotes are not monophyletic (yeast external).
Fungi Mammalia Bacillariophyta Amphibia Viridaeplanatae 16s rRNA – 20 Species Secondary structure based tree
Archaea Eukarya Bacteria 16s rRNA –91 Species Secondary structure based tree
Collins et al., 2000 • After completing this project, we discovered a related, earlier work from David Penny’s group. When determining evolutionary relationships between some catalytic RNA molecules, they constructed a 16S rRNA tree based on a similar “distance approach”. • We compared our results to • the trees published in their article • (using a different distance algorithm, • RNAdistance, by Shapiro & Zhang).
Collins’ 16s r RNA secondary struct based tree Collins et al., 2000. Archaea Collins’ 16s rRNA sequence based tree 16 Species Archaea Bacteria Bacteria
Archaea Bacteria Our Tree, 13 Out of 16 Collins’ Species Secondary structure based tree
A Close Look at the Trees Collins’ 16s rRNA seq based tree outgroups Our 16s second. struct. tree Collins’ 16s second. struct. based tree
Methanobacteruim Methanococcus Thermoplasma A Close Look at Sec. Strucs. Supports a “Thermoplasma Outgroup” Theory
Conclusions • Encouraging results • Accuracy of structure based trees is comparable to sequence based trees. • Warning: Reliable secondary structures • are crucial for accurate tree reconstruction.