Download
1 / 14
160 likes | 539 Views
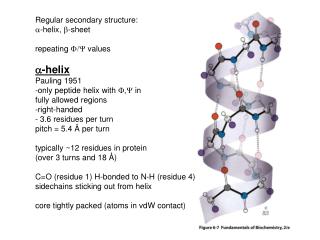
Regular secondary structure: a -helix, b -sheet repeating F/Y values a -helix Pauling 1951 -only peptide helix with F,Y in fully allowed regions -right-handed - 3.6 residues per turn pitch = 5.4 Å per turn typically ~12 residues in protein (over 3 turns and 18 Å )

E N D
Regular secondary structure: a-helix, b-sheet repeating F/Y values a-helix Pauling 1951 -only peptide helix with F,Y in fully allowed regions -right-handed - 3.6 residues per turn pitch = 5.4 Å per turn typically ~12 residues in protein (over 3 turns and 18 Å) C=O (residue 1) H-bonded to N-H (residue 4) sidechains sticking out from helix core tightly packed (atoms in vdW contact)
b-sheet also uses full H-bonding capacity of backbone (between neighboring chains) anti-parallel or parallel orientation not exactly completely extended (would put sidechains pointing at neighboring polypeptide and give a steric clash) Observed F=-90 - -180 observed Y = 150-180 -pleated or rippled sheet pattern -overy other sidechain points up -better geometry for C=O…H-N hydrogen bond sheets 2-22 strands, ave.=6, up to 15 residues (small also observed) right-handed twist (compromise between L-amino acid centers and maximizing interchain H-bonding)
Collagen, a triple helix -most abundant vertebrate protein!!! strong, stress-bearing fibers components of bone, teeth cartilage, tendon, and fibrous matrices of skin and blood vessels 3 polypeptide chains, distinctive a.a. composition: 33% gly 15-30% pro, hyp 4-hydroxyprolyl residue 3-hydroxyprolyl and 5-hydroxylysyl also occur smaller amounts Created by modification of synthesized polypeptide chains Pro Hyp prolyl hydroxylase (requires ascorbic acid for its activity) Scurvey…..limeys….. 2° structure? Proline!!!!!
Collagen has G-X-Y repeating sequence X-often pro, Y-often Hyp Proline cannot form a-helices (f=-60°, restricted, and no N-H for H-bond to C=O) So, collagen forms gentle left-handed helix with 3 residues per turn And, 3 helices reverse the twist to form a right-handed 3-helical coil
Every 3rd residue through center of triple helix (so crowded that only room for gly sidechain…explains absolute requirement for gly every 3rd residue) 3 chains staggered, so only gly in center of coil Each gly N-H forms strong H-bond with Pro C=O on neighboring chain to stabilize overall structure Pro, Hyp bulky & inflexible, making assembly rigid Collagen’s strength comes from: well-packed, rigid, triple helix (can’t unwind) Crosslinking: lysyl oxidase, then: 2X allysine, aldol, His adds, 5-hydroxylysine, increases with age…tough old meat…
Non-repetitive protein structure Mostly globular…several types of regular secondary struc (a,b,turns), irregular and unique also possible (non-repetitive F,Y values) Don’t confuse with random coil (what I have been calling spaghetti in class) #3 often Gly (no R group) Clash! #2 often Pro (-60°)
Random coil: disordered and rapidly fluctuating conformation assumed by denatured (fully unfolded) proteins (imagine hydrogen bond acceptors and donors making all H-bonds with water, hydrophobics randomly burying surface with self and non-self polypeptide chains). • Irregular Structures: in native proteins, irregular structures are no less ordered than helices or b-sheets, simply more difficult to describe • Distortions: a.a. sequence variations, overall structure of folded protein can distort regular conformations of secondary structural elements. -1st and last turns of helix (don’t have all H-bonds) -helix capping (Gln/Asn sidechain folds back to H-bond with backbone exposed C=O groups…) -b-bulge (1 residue in b-strand not H-bonded…pokes out) -Pro kinks helices & sheets -(ii+3,4 steric clashes big sidechains in a helix) Propensities of a.a. in known structures are useful to predict 2° structure (Pro,Gly in turns, between alpha/beta structures)
Protein structure determination Protein Data Bank (www.rcsb.org), ~30,000 structures, downloadable coordinate files X-ray crystallography (direct imaging of molecules) From optical principles, error associated with locating an object is on the order of the wavelength of light used to observe it (covalent bonds and X-rays both ~1.5 Å) visible light ~4000 Å, too long… Crystals diffract X-rays onto a radiation counter (or photographic film…) X-rays now normally produced from a particle accelerator, called a synchrotron ( Grenoble, Los Alamos, huge national user facilities…). Intensities (darkness of spots) used to calculate mathematically the position of electrons that diffracted the X-ray. X-rays interact primarily with electrons, not nuclei. Therefore, crystallographers plot electron density maps and then try to model protein residues into the shape of the map.
Every ( e.g. Tyrosine 56 beta carbon) atom in the lattice of protein molecules deflects the incident X-ray with the same angle… You don’t have to know how diffraction works, only that we can reconstruct electron density from the diffraction pattern.
X-ray, cont’d.1 Even though structures presented as atomic models, most structures are less than atomic resolution!!!!! -Proteins arranged as repeating, 3-dimensional lattices -Protein crystals are 40-60% water (must be in aqueous conditions for their structural integrity…therefore protein crystals are soft, jellylike in consistency) -Molecules in crystal are typically disordered by 1 Å or more -Resolution is typically 1.5-3.0Å, but some more ordered (better resolution) and some worse…
(1b) Accuracy and feasibility of crystal structure analysis depends on resolution -Trace distinctive backbone, deduce orientations of sidechains, but Ile, Leu, Thr and Val hard to distinguish. -Hydrogen only observed at less than 1.2 Å, and N and O are hard to distinguish *Utilize primary sequence to fit it into the density C-C bond length ~1.5 Å Model hydrogen positions and N/O identity based on nearby groups Can lead to bias towards ideal, “normal” geometry and H- bonding clearly shows atoms
X-ray, cont’d.2 • Crystalline proteins mostly in native conformation • Protein basically solvated by crystallization solution except where it contacts • neighboring protein molecules (small patches called crystal contacts) (40-60% • water is similar to a living cell) • Proteins can be crystallized in more than one crystal form, and still give the same • structure…your book claims that NMR vs. crystal comparison shows proteins with • same structure typically (BUT, there can be important differences!) • Many enzymes are catalytically active in the crystalline state, and since they must • have their catalytic sidechains perfectly oriented, this is strong evidence of its • occurrence in the crystal • Disordered portions of proteins (loops, termini, unfolded elements) do not diffract • X-rays, because the position of the atoms in each molecule in the crystal is not the same. Therefore, these portions in an otherwise ordered crystal give no structural information and are left out of structure models.