Download

1 / 40
650 likes | 1.55k Views
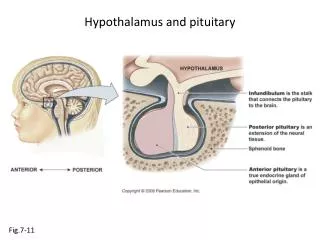
DISORDERS OF THE HYPOTHALAMUS AND PITUITARY. Hormones of the Pituitary Gland. Pituitary: The Master Gland. The pituitary gland is located at the base of the skull in a saddle-shaped cavity of the sphenoid bone: the sella turcica It is connected to the hypothalamus by the pituitary stalk.

E N D
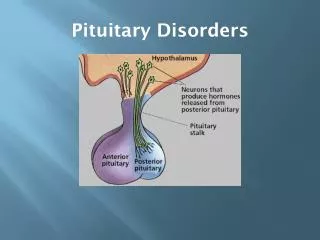
Pituitary: The Master Gland • The pituitary gland is located at the base of the skull in a saddle-shaped cavity of the sphenoid bone: the sellaturcica • It is connected to the hypothalamus by the pituitary stalk. • It is composed of an anterior (adenohypophysis) and a posterior (neurohypophysis) lobe • The anterior pituitary gland originates from the Rathke pouch as an invagination of the oral ectoderm • major regulator of an elaborate hormonal system • receives signals from the hypothalamus and responds by sending pituitary hormones to target glands • The target glands produce hormones that provide negative feedback at the level of the hypothalamus and pituitary • This feedback mechanism enables the pituitary to regulate the amount of hormone released into the bloodstream by the target glands.
Vascular Supply of the Pituitary • Originates from the internal carotid via hypophysealarteries. • This network of vessels forms a unique portal circulation connecting the hypothalamus and pituitary. • It is through this portal venous system that hypothalamic hormones are delivered to the anterior pituitary gland. • Anterior pituitary hormones, in turn, are secreted into a secondary plexus of portal veins that drain into the dural venous sinuses.
Anterior Pituitary Cell Types • Somatotropes • Growth hormone (GH) • Lactotropes • Prolactin (PRL) • Thyrotropes • Thyroid-stimulating hormone (TSH) • Corticotropes • Pro-opiomelanocortin(POMC), the precursor of adrenocorticotropic hormone (ACTH) • Gonadotropes • Luteinizing hormone (LH) • Follicle-stimulating hormone (FSH)
Growth Hormone • GH is secreted in a pulsatile fashion under the regulation of hypothalamic hormones • The biologic effects of GH include increases in linear growth, bone thickness, soft tissue growth, protein synthesis, fatty acid release from adipose tissue, insulin resistance, and blood glucose levels.
Prolactin • The primary physiologic role is the initiation and maintenance of lactation. • Prepares the breasts for lactation and stimulates milk production postpartum. • During pregnancy, PRL stimulates the development of the milk secretory apparatus, but lactation does not occur because of the high levels of estrogen and progesterone. • After delivery, the estrogen and progesterone levels drop and physiologic stimuli such as suckling and nipple stimulation signal PRL release and initiate lactation.
Thyroid-Stimulating Hormone • stimulates release of thyroxine (T4) and triiodothyronine (T3) from the thyroid gland through the formation of cyclic adenosine monophosphate (cAMP) and the G-protein second messenger system. • Deficiency of TSH results in inactivity and atrophy of the thyroid gland, whereas excess TSH results in hypertrophy and hyperplasia of the thyroid gland.
Adrenocorticotropic Hormone • secreted in a diurnal pattern. • It acts on the adrenal cortex to stimulate cortisol synthesis and secretion. • ACTH and cortisol levels are highest in the morning at the time of waking, are low in the late afternoon and evening, and reach their nadir 1–2 hr after the beginning of sleep. • ACTH also appears to be the principal pigmentary hormone in humans. • Physiologic conditions such as stress, fasting, and hypoglycemiastimulate release of CRH and ACTH.
Luteinizing Hormone and Follicle-Stimulating Hormone • FSH Stimulates follicular development in the ovary and of gametogenesis in the testis. • LH promotes luteinization of the ovary and Leydig cell function of the testis. • Secretion of LH is inhibited by androgens and estrogens, and secretion of FSH is suppressed by gonadal production of inhibin
Posterior Pituitary Cell Types • Arginine vasopressin (AVP; antidiuretic hormone [ADH] • regulates water conservation at the level of the kidney by increasing the permeability of the renal collecting duct to water. • Oxytocin • stimulates uterine contractions at the time of labor and delivery in response to distention of the reproductive tract, and it stimulates smooth muscle contraction in the breast during suckling, which results in milk let-down.
Hypopituitarism • Denotes underproduction of growth hormone (GH) alone or in combination with deficiencies of other pituitary hormones. • incidence is between 1 in 4,000 and 1 in 10,000 live births. • Isolated GH deficiency (IGHD) is caused by abnormalities of the GHRH receptor, growth hormone genes, and by genes located on the X chromosome
Congenital Hypopituitarism • usually of normal size and weight at birth although those with MPHD and genetic defects have birth lengths that average 1 SD below the mean. • Children with severe defects in GH production or action are more than 4 SD below the mean • Delayed closure of the epiphyses permits growth beyond the normal age when growth should be complete. • Usually present with neonatal emergencies such as apnea, cyanosis, or severe hypoglycemia with or without seizures. Microphallus in boys provides an additional diagnostic clue. • Prolonged neonatal jaundice is common
Acquired Hypopituitarism • The child initially is normal • manifestations gradually appear and progress • Signs of pituitary insufficiency • loss of weight, asthenia, sensitivity to cold, mental torpor, and absence of sweating • Sexual maturation fails to take place or regresses if already present, amenorrhea and loss of pubic and axillary hair. • Diabetes insipidus may be present early but tends to improve spontaneously as the anterior pituitary is progressively destroyed. • If the lesion is an expanding tumor • headache, vomiting, visual disturbances, pathologic sleep patterns, decreased school performance, seizures, polyuria, and growth failure may occur. • In children with craniopharyngiomas, visual field defects, optic atrophy, papilledema, and cranial nerve palsy are common.
Diagnosis of GH Deficiency • Absent or low levels of GH in response to stimulation. • Provocative tests include administration of insulin, arginine, clonidine, or glucagon. • Chronic GH deficiency • poor linear growth, delayed skeletal age • peak levels of GH (<10 ng/mL) in each of 2 provocative tests • Acute GH deficiency, • a high clinical suspicion of GH deficiency • peak levels of GH (<10 ng/mL) in each of 2 provocative tests • Levels of TSH, thyroxine (T4), ACTH, cortisol, gonadotropins, and gonadal steroids may provide evidence of other pituitary hormonal deficiencies.
Imaging Techniques • CT is appropriate for recognizing suprasellar calcification associated with craniopharyngiomas and bony erosions accompanying histiocytosis • MRI provides a much more detailed view of hypothalamic and pituitary anatomy • Craniopharyngiomasare common and pituitary adenomas are rare in children with hypopituitarism • Skeletal maturation is delayed in patients with IGHD and may be even more delayed when there is combined GH and TSH deficiency. • Dual photon x-ray absorptiometry shows deficient bone mineralization, deficiencies in lean body mass, and a corresponding increase in adiposity.
Differential Diagnosis CONSTITUTIONAL GROWTH DELAY • a common variant of normal growth • Length and weight measurements are normal at birthand for the first 4–12 mo of life • Height is sustained at a lower percentile during childhood. • The pubertal growth spurt is delayed • Family history of short stature in childhood, delayed puberty, and eventual normal stature. • cause is thought to be persistence of the relatively hypogonadotropic state of childhood
Differential Diagnosis PSYCHOSOCIAL DWARFISM • Emotional deprivation is an important cause of retardation of growth and mimics hypopituitarism. • The condition is known as psychosocial dwarfism, maternal deprivation dwarfism, or hyperphagic short stature. • Puberty may be normal or even premature in its appearance. • Proof may be difficult to establish because the parents or caregivers often hide the true family situation from professionals, and the children rarely divulge their plight. • frequently have perverted or voracious appetites, enuresis, encopresis, insomnia, crying spasms, and sudden tantrums. • The subgroup of children with hyperphagia and a normal body mass index tends to show catch-up growth when placed in a less stressful environment.
Treatment • classic GH deficiency • hGHis 0.18–0.3 mg/kg/wk during childhood. • Recombinant GH is administered subcutaneously in 6 or 7 divided doses. • Maximal response to GH occurs in the 1st year of treatment. • Growth hormone therapy should be continued until near final height is achieved. Criteria for stopping treatment include a decision by the patient that he or she is tall enough, a growth rate less than 1 inch/yr and a bone age >14 yr in girls and >16 yr in boys. • Some patients develop either primary or central hypothyroidism while under treatment with GH. Similarly, there is a risk of developing adrenal insufficiency. • In children with MPHD, replacement should also be directed at other hormonal deficiencies.
Adverse Effects of hGH Treatment • Some patients treated with GH have developed leukemia • Growth hormone treatment does not increase the risk for recurrence of brain tumors or leukemia. • Other reported side effects include pseudotumorcerebri, slipped capital femoral epiphysis, gynecomastia, and worsening of scoliosis. • Fasting and postprandial insulin levels are characteristically low before treatment, and they normalize during GH replacement. Treatment does not increase risk of type 1 diabetes, but it may increase the risk of type 2 diabetes.
Diabetes Insipidus • polyuria and polydipsia • Central DI • vasopressin deficiency • Nephrogenic DI • vasopressin insensitivity at the level of the kidney
Evaluation of a Child with Polyuria and Polydipsia • The cause of pathologic polyuria or polydipsia (exceeding 2 L/m2/24 hr) may be difficult to establish in children. • Infants may present with irritability, failure to thrive, and intermittent fever. • Careful history which should quantify the child's daily fluid intake and output and establish the voiding pattern, nocturia, and primary or secondary enuresis. • A complete physical examination should establish the patient's hydration status, and the physician should search for evidence of visual and central nervous system dysfunction as well as other pituitary hormone deficiencies. • Laboratory work up • Serum for osmolality, sodium, potassium, blood urea nitrogen, creatinine, glucose, and calcium • Urine for osmolality, specific gravity, and glucose determination
Evaluation of a Child with Polyuria and Polydipsia • The diagnosis of DI is established if • serum osmolality is greater than 300 mOsm/kg • urine osmolality is less than 300 mOsm/kg • DI is unlikely if the serum osmolality is less than 270 mOsm/kg or the urine osmolality is greater than 600 mOsm/kg. • If the patient's serum osmolality is less than 300 mOsm/kg (but greater than 270 mOsm/kg) and pathologic polyuria and polydipsia are present, a water deprivation test is indicated to establish the diagnosis of DI and to differentiate central from nephrogenic causes.
Evaluation of a Child with Polyuria and Polydipsia • In the inpatient post-neurosurgical setting, central DI is likely if hyperosmolality (serum osmolality >300 mOsm/kg) is associated with urine osmolality less than serum osmolality. • It is important to distinguish between polyuria resulting from postsurgical central DI and polyuria resulting from the normal diuresis of fluids received intraoperatively. Both cases may be associated with a large volume (>200 mL/m2/h) of dilute urine, although in patients with DI, the serum osmolality is high in comparison with patients undergoing postoperative diuresis.
Central Diabetes Insipidus • Can result from multiple etiologies • Genetic mutations in the vasopressin gene • Trauma (accidental or surgical) to vasopressin neurons • Congenital malformations of the hypothalamus or pituitary • Neoplasms; infiltrative, autoimmune • Infectious diseases affecting vasopressin neurons or fibertracts • Increased metabolism of vasopressin • Autosomal dominant central DI usually presents within the first 5 yr of life and results from mutations in the vasopressin gene.
Treatment of Central DI • Fluid Therapy • With an intact thirst mechanism and free access to oral fluids, a person with complete DI can maintain plasma osmolality and sodium in the high normal range • Vasopressin Analogs. • dDAVP(desmopressin) available in an intranasal preparation (onset 5–10 min) and as tablets (onset 15–30 min) • Aqueous Vasopressin • Central DI of acute onset following neurosurgery is best managed with continuous administration of synthetic aqueous vasopressin (pitressin).
Nephrogenic Diabetes Insipidus • Genetic nephrogenic DI • Usually presents within the first several wk of life but may only become apparent after weaning or with longer periods of nighttime sleep. • fever, vomiting, and dehydration, failure to thrive • Long-standing ingestion and excretion of large volumes of water may lead to nonobstructivehydronephrosis, hydroureter, and megabladder. • Acquired nephrogenic DI • may result from hypercalcemia or hypokalemia and is associated with the following drugs: lithium, demeclocycline, foscarnet, clozapine, amphotericin, methicillin, and rifampin. • Impaired renal concentrating ability can also be seen with ureteral obstruction, chronic renal failure, polycystic kidney disease, medullary cystic disease, Sjögren syndrome, and sickle cell disease.
Treatment of Nephrogenic DI • Focuses on elimination, if possible, of the underlying disorder, such as offending drugs, hypercalcemia, hypokalemia, or ureteral obstruction. • Foods with the highest ratio of caloric content to osmotic load (Na <1 mmol/kg/24 hr) should be ingested to maximize growth and to minimize the urine volume required to excrete the solute load. Even with the early institution of therapy, however, growth failure and mental retardation are common. • Thiazide diuretics • decrease the overall urine output • induce a state of mild volume depletion by enhancing sodium excretion at the expense of water and by causing a decrease in the glomerular filtration rate, which results in proximal tubular sodium and water reabsorption.
Other Abnormalities of Arginine Vasopressin Metabolism and Action
Hyponatremia • serum sodium <130 mEq/L • Usually associated with severe systemic disorders and is most often due to • (1) intravascular volume depletion • (2) excessive salt loss • (3) hypotonic fluid overload • The syndrome of inappropriate antidiuretic hormone (SIADH) is an uncommon cause of hyponatremia in children.
Clinical Parameters to Distinguish Between SIADH, Cerebral Salt Wasting and Central DI
Hyperpituitarism • Primary hypersecretion of pituitary hormones occurs rarely in children. When it does, it usually occurs as a result of a pituitary adenoma. Primary hyperpituitarism should not be confused with secondary hyperpituitarism, which occurs in the setting of target hormone deficiencies resulting in decreased hormonal feedback, such as in hypogonadism, hypoadrenalism, or hypothyroidism. • The most frequently seen adenoma during childhood • Prolactinoma • Corticotropinoma • Somatotropinoma
PROLACTINOMA • Prolactin-secreting pituitary adenomas are the most common tumors of the pituitary in adolescents • With the advent of MRI, more of these tumors, particularly microadenomas (<1 cm), are being detected. • The most common presenting manifestations are headache, amenorrhea, and galactorrhea. • The disorder affects more than twice as many girls as boys; most patients have undergone normal puberty before becoming symptomatic. Only a few have delayed puberty. In some kindreds with type I multiple endocrine neoplasia (MEN), prolactinomas are the presenting feature during adolescence. • Prolactin levels may be moderately (40–50 ng/mL) or markedly (10,000–15,000 ng/mL) elevated. • Most prolactinomas in children are large (macroadenomas), cause the sella to enlarge, and in some cases cause visual field defects. • Treatment for most children has been surgical resection by transfrontal or transsphenoidal approach. Prolactinoma can also be effectively managed medically in most patients by treatment with bromocriptine or long-acting cabergoline. About 80% of adult patients respond with shrinkage of the tumor and marked decreases in serum prolactin levels.
Corticotropinoma • In pediatrics, corticotropinomas are the most common adenomas seen prepubertally although they occur at all ages. • Cushing's disease refers specifically to an ACTH-producing pituitary adenoma that stimulates excess cortisol secretion. • Adenomas causing Cushing's disease are significantly smaller than all other types of adenomas at presentation. • The most sensitive indicator of excess glucocorticoid secretion in children is growth failure, which generally precedes other manifestations. Patients develop weight gain that tends to be centripetal rather than generalized. Pubertal arrest, fatigue, and depression are also common. • Transsphenoidal surgery is the treatment of choice for Cushing's disease in children. Initial remission rates of 70–98% of patients and long-term success rates of 50–98% have been reported. Residual transient hypoadrenalism is often observed after surgery, lasting as long as 30 months.
Excess Growth Hormone Secretion and Pituitary Gigantism • In young persons with open epiphyses, overproduction of GH results in gigantism; in persons with closed epiphyses, the result is acromegaly. Often, some acromegalic features are seen with gigantism, even in children and adolescents. After closure of the epiphyses, the acromegalic features become more prominent. • Pituitary gigantism is rare, and its cause is most often a pituitary adenoma • The cardinal clinical feature of gigantism is longitudinal growth acceleration secondary to GH excess. • The usual manifestations consist of coarse facial features and enlarging hands and feet • Acromegalicfeatures consist chiefly of enlargement of the distal parts of the body, but manifestations of abnormal growth involve all portions • The circumference of the skull increases, the nose becomes broad, and the tongue is often enlarged, with coarsening of the facial features. The mandible grows excessively, and the teeth become separated. Visual field defects and neurologic abnormalities are common; signs of increased intracranial pressure appear later. The fingers and toes grow chiefly in thickness.
Diagnosis of Growth Hormone Excess • The gold standard for making the diagnosis of GH excess is the failure to suppress serum GH levels to less than 5 ng/dL after a 1.75 g/kg oral glucose challenge (maximum, 75 g). • This test measures the ability of IGF-I to suppress GH secretion because the glucose load results in insulin secretion, leading to suppression of IGFBP-I, which results in an acute increase in free IGF-I levels. The increased free IGF-I suppresses GH secretion within 30–90 min. This test can be abnormal in diabetic patients. • A single measurement of GH is inadequate because GH is secreted in a pulsatile manner. Therefore, the use of a random GH measurement can lead to both false-positive and false-negative results.
Treatment • The goals of therapy are to remove or shrink the pituitary mass, to restore GH and secretory patterns to normal, to restore IGF-I and IGFBP-3 levels to normal, to retain the normal pituitary secretion of other hormones, and to prevent recurrence of disease. • For well-circumscribed pituitary adenomas, transsphenoidal surgery is the treatment of choice and may be curative. The tumor should be removed completely. • Radiotherapy is recommended if GH hypersecretion is not normalized by surgery. • Medical therapy • Octreotide