Download

1 / 34
490 likes | 2.18k Views
Sclerosi Laterale Amiotrofica. Prof. Gabriele Siciliano Dipartimento di Neuroscienze, Facoltà di Medicina e Chirurgia, Pisa. Sclerosi Laterale Amiotrofica. atrofia gliotica. S clerosi. cordoni laterali del midollo spinale. L aterale. riduzione della massa muscolare. A miotrofica.

E N D
Sclerosi Laterale Amiotrofica Prof. Gabriele Siciliano Dipartimento di Neuroscienze, Facoltà di Medicina e Chirurgia, Pisa
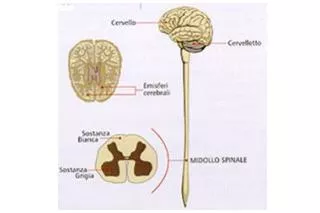
atrofia gliotica Sclerosi cordoni laterali del midollo spinale Laterale riduzione della massa muscolare Amiotrofica Sclerosi Laterale Amiotrofica (SLA) La più grave fra le malattie che colpiscono il motoneurone:
Malattie del motoneurone: classificazione • Forme idiopatiche- sclerosi laterale amiotrofica; - paralisi bulbare progressiva;- atrofia muscolare progressiva;- sclerosi laterale primaria;- sclerosi laterale amiotrofica familiare;- sclerosi laterale amiotrofica giovanile;- malattia del motoneurone di Madras;- malattia del motoneurone monomielica. • 2. Forme tossiche– latirismo;– Konzo;- SLA dell'isola di Guam.
SLA: corteccia cerebrale motoria SLA normale
SLA: definizione • La SLA è una malattia neurodegenerativa caratterizzata dalla progressiva perdita dei motoneuroni: • corticali (1°motoneurone); • spinali (2°motoneurone).
Caratteristiche cliniche della SLA • Il primo motoneurone origina dal quinto strato della corteccia motoria, discende attraverso la via corticospinale e corticobulbare e termina in sinapsi con il secondo motoneurone; • Il secondo motoneurone è rappresentato dalla cellula delle corna anteriori del midollo spinale e dal suo omologo nel tronco encefalico.
Selettività nella degenerazione • Risparmio dei: • sistemi sensitivi; • sistemi della coordinazione motoria. • La selettività interessa anche il sistema motorio, infatti non sono mai colpiti: • i motoneuroni che controllano la motilità oculare; • i neuroni motori degli sfinteri striati anale ed uretrale, localizzati nel midollo spinale sacrale, nucleo di Onuf, a livello S2-S4.
Epidemiologia • La SLA colpisce circa 1-3 persone per 100.000 per anno. • La prevalenza è di 5-9 casi su 100.000, valore dipendente dal tempo di sopravvivenza della malattia. • Rapporto maschi/femmine= 2/1 • È stimato che ad ogni momento siano presenti circa 20000 persone affette da SLA negli Stati Uniti, 28000 nell'Unione europea (3000 in Italia). • Esordio primi sintomi in media tra 55 e 65 anni.
FORME EZIOPATOGENETICHE • SLA SPORADICA o MALATTIA DI CHARCOT; • SLA FAMILIARE.
SLA sporadica: ipotesi eziopatogenetiche • ECCITOTOSSICITÀ GLUTAMMATERGICA Il glutammato determina un aumento dell’afflusso di Ca++ intracellulare che si traduce in un’attivazione enzimatica (endonucleasi, fosfolipasi, ossido nitrico sintetasi) che culmina nella degenerazione e necrosi cellulare.
SLA sporadica: ipotesi eziopatogenetiche • STRESS OSSIDATIVO: • aumentata attività della selenioproteina enzimatica glutatione perossidasi nel midollo spinale; • aumentato mRNA per la SOD1 in motoneuroni residui; • aumentato livello nel midollo spinale e nella corteccia frontale di carbonili proteici derivati dall’ossidazione di alcune proteine.
SLA familiare • Rappresenta circa il 10% dei casi SLA; • Trasmissione autosomico dominante; • In 1/5 dei casi è stata individuata una mutazione nel gene che codifica per l’enzima Cu++/Zn++ superossidodismutasi (SOD1) localizzato nel cromosoma 21.
SLA: classificazione clinica E’ possibile classificare la SLA in relazione: • al coinvolgimento del 1° e/o del 2° motoneurone; oppure • sulla base del distretto corporeo maggiormente colpito all’esordio della malattia.
SLA: caratteristiche cliniche • Motoneurone corticale (1°MN): • deficit di forza; • spasticità; • labilità emotiva; • ROT vivaci; • Babinski positivo.
SLA: caratteristiche cliniche • Motoneurone spinale (2°MN) • atrofia; • fascicolazioni; • ROT ridotti/assenti; • crampi; • disfagia; • disartria; • insufficienza respiratoria.
SLA: forme cliniche Forma Comune (45%-50%) Esordio insidioso con una progressiva riduzione di forza alle mani, dove sono in un primo momento interessati gli estensori piuttosto che i flessori. ATROFIA Mano di scimmia Mano di Aran-Duchenne L’atrofia si diffonde anche agli avambracci e talora ai muscoli della spalla e poi agli arti inferiori.
SLA:forme cliniche Forma Bulbare (25%) • I primi sintomi sono usualmente: • difficoltà a pronunciare fonemi: • consonantici; • labiali; • linguali. • ipotrofia e fascicolazioni alla lingua.
SLA:forma bulbare • In fase più avanzata: • protrusione della lingua impossibile; • voce nasale; • disartria e/o anartria; • ridotta motilità del velo palatino; • difficoltà della deglutizione inizialmente per i liquidi, fino alla totale incapacità a deglutire;
SLA:forma bulbare In fase più avanzata: • compromissione delle corde vocali con disfonia fino all’afonia; • compromissione della funzione respiratoria; • difficoltà nella tosse; • sonno disturbato; • ridotta capacità vitale forzata, dato prognosticamente negativo predittivo di una ridotta aspettativa di vita.
Danno muscolare nella SLA La perdita dei motoneuroni bulbari determina l’insorgenza di disfagia, disfonia, disartria e dispnea La perdita dei motoneuroni spinali determina deficit della muscolatura respiratoria INSUFFICIENZA RESPIRATORIA Lingua Mammellonata in paziente con SLA (interessamento del nervo ipoglosso, XII paio di nervi cranici)
SLA: forme cliniche Forma Pseudopolinevritica (25%-30%) E’ caratterizzata da un deficit motorio ai muscoli della loggia antero-esterna della gamba, talora unilaterale, ma in breve tempo bilaterale. Il soggetto inciampa per caduta del piede, oppure se il disturbo ha inizio prossimale, ha difficoltà a sollevarsi dalla posizione seduta. Il disturbo motorio appare in genere prima dell’atrofia. I riflessi profondi sono in un primo tempo ridotti o aboliti e i segni piramidali sono tardivi.
Progressione • Il riconoscimento della velocità di progressione nella storia naturale della malattia è un importante dato anche in senso diagnostico. • Un inizio agli arti è presente nel 65-80% dei casi, inizio bulbare nel 20-25%; • La malattia quindi interessa altre regioni, più frequentemente verso segmenti muscolari contigui; • La progressiva perdita delle cellule delle corna anteriori risulta in una progressiva disabilità, che in ultima analisi confina il paziente a letto; • Il decesso avviene entro 3 anni nel 50% dei casi.
SLA: gradi di certezza diagnostica • DEFINITA • disfunzione 1° e 2° MN in 3 distretti. • PROBABILE • disfunzione 1° e 2° MN in 2 distretti. • POSSIBILE • disdfunzione 1° MN + 2° MN in un distretto (arto superiore, arto inferiore, tronco o faccia-faringe); • oppure disfunzione 1° MN in 2 o 3 distretti. • PROBABILE SUPPORTATA DA ESAMI DI LABORATORIO • basata su criteri clinici analoghi a quelli della SLA clinicamente possibile, ma con la presenza di segni elettromiografici di coinvolgimento del secondo motoneurone in almeno 2 estremità .
SLA: diagnosi differenziale • neuropatie periferiche; • miopatie; • malattie infettive del SNC; • cerebropatie vascolari ischemiche; • atrofia muscolare spinale; • sclerosi multipla; • miastenia grave; • deficit di esosaminidasi A; • tireotossicosi; • neoplasie; • intossicazione da piombo; • lesioni spinali.
Fascicolazioni Riduzione delle unità motorie: A-lieve, non specificaB-moderata riduzioneC-severa riduzione
Il riluzolo inibisce il rilascio del Glutammato. Glutammato Riluzolo Canali del sodio Il riluzolo blocca i canali del sodio voltaggio dipendenti. Trattamento specifico della malattia: riluzolo Solo il Riluzolo, un antagonista del glutammato, è in grado di prolungare di 3-6 mesi la sopravvivenza.
Riluzolo • E’ il solo farmaco approvato dalla Food and Drug Administration per il trattamento della SLA; • i pazienti trattati con riluzolo rimangono negli stadi di malattia più lievi più a lungo rispetto ai controlli; • efficacia del riluzolo nel: • prolungare la sopravvivenza nella SLA; • nel ritardare il ricorso ad interventi di sostegno alla sopravvivenza quali la tracheotomia e la ventilazione meccanica.
SLA: terapia sintomatica • Antidepressivi triciclici, attivi contro la scialorrea; • Carbamazepina, per le fascicolazioni e i crampi; • Baclofen, contro la spasticità, • Gabapentin, contro il dolore neuropatico; • Gastrostomia percutanea endoscopica (PEG); • Ventilazione non invasiva; • Tracheotomia.
SLA: prospettive terapeutiche • Farmaci neurotrofici e gliotrofici; • Farmaci inibitori dell’apoptosi; • Farmaci protettori nei confronti dello stress ossidativo; • Terapia con cellule staminali.
SLA: link • http://www.aisla.it/ • http://www.wfnals.org/ • http://neuro.wustl.edu/neuromuscular/