Understanding Polymyositis and Dermatomyositis: Clinical Insights and Management
521 likes | 833 Views
Polymyositis (PM) and dermatomyositis (DM) are important inflammatory myopathies characterized by acquired muscle weakness. PM typically presents with symmetrical, proximal muscle weakness without skin involvement, while DM features similar weakness alongside distinctive rashes. Both conditions can be linked to systemic autoimmune diseases. Diagnosis involves clinical assessment, serum enzyme tests, and muscle biopsies. The management often requires immunosuppressive therapies. Understanding these conditions is crucial for timely intervention and improving patient outcomes.

Understanding Polymyositis and Dermatomyositis: Clinical Insights and Management
E N D
Presentation Transcript
Polymyositis , Dermatomyositis By DrP.Harischandra 28 Sept 2015
Introduction • The inflammatory myopathies represent the largest group of acquired and potentially treatable causes of skeletal muscle weakness. They are classified into three major groups: polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM). • Myopathies is 1 in 100,000 • PM rare, DM affects both children and adults, F>M • IBM M>F, W>B > 50 yrs. of age
Classification • Group I Idiopathic polymyositis • Group II Idiopathic dermatomyositis • Group III Dermatomyositis (polymyositis) associated with neoplasia • Group IV Childhood dermatomyositis associated with vasculitis • Group V Polymyositis with collagen vascular disease
Clinical features • Progressive and symmetric muscle weakness except for IBM, which can have an asymmetric pattern • Increasing difficulty with everyday tasks requiring the use of proximal muscles, such as getting up from a chair, climbing steps, stepping onto a curb, lifting objects, or combing hair • Fine-motor movements –late PM/DM. Early early in IBM • Ocular muscles not involved – diagnosis questioned
Neck muscles involved- dysphagia, head drop • Late stages- respiratory muscles • Sensory system normal • Reflexes – preserved, May be absent in Severe atrophy of muscles or in advanced IBM • Myalgia and muscle tenderness may occur –DM • Weakness in PM and DM progresses sub acutely over a period of weeks or months • IBM progresses very slowly, over years, simulating a late-life muscular dystrophy
Polymyositis PM • Actual onset of PM is often not easily determined, usually late presentation to medic-contrast to DM where rash facilitates early recognition • PM mimics many other myopathies and is a diagnosis of exclusion • subacute inflammatory myopathy affecting adults, and rarely children • No h/o exposure to myotoxic drugs or toxins, endocrinopathy, neurogenic disease, muscular dystrophy, biochemical muscle disorder
Isolated entity, PM is a rare (and over diagnosed) disorder; more commonly, PM occurs in association with a systemic autoimmune or connective tissue disease • Drugs, especially d-penicillamine, statins, or zidovudine (AZT), may also trigger an inflammatory myopathy similar to PM
Polymyositis • Symmetric proximal muscle weakness • Elevated serum muscle enzymes* • CK, CK-MB, AST, ALT, LD, Aldolase • Myopathic changes on EMG • Muscle biopsy • cellular infiltrate is predominantly within the fascicle with inflammatory cells invading individual muscle fibers • cell–mediated, increased numbers of cytotoxic CD8+ T cells, which appear to recognize an antigen on the muscle fiber surface
Dermatomyositis • Dermatomyositis(DM) is a rare autoimmune disorder characterized by inflammation of striated muscle, causing proximal muscle weakness with skin involvement.
Epidemiology and Etiology • Incidence is about 2-10/million population per annum • It occurs in all races and at all ages • M:F ratio is 1:3 • Etiology is unknown, although viruses(e.g. Coxsackie, rubella, influenza) have been implicated • Persons with HLA-B8/DR3 appear to be predisposed
Dermatomyositis • characteristic rash accompanying, or more often preceding, muscle weakness • The rash may consist of a blue-purple discoloration on the upper eyelids with edema -a flat red rash on the face and upper trunk, and erythema of the knuckles with a raised violaceous scaly eruption (Gottron'ssign • knees, elbows, malleoli, neck and anterior chest (often in a V sign), or back and shoulders (shawl sign)-may worsen after sun exposure
Clinical Presentation • PC: rash, general weakness, multiple sores, insomnia – 6/12 • HPC: Pt had been well until the Sxbegan and progressed gradually until she presented at the hospital • ODQ: Wt loss+, Easy fatigability+, dizzinesso • SE: cougho, chest paino, palpitationso, dysphagia+(with solid food), odynophagia+, vomitingo, diarrheao, dysuriao, frequencyo, headacheo
Dilated capillary loops at the base of the fingernails are also characteristic • cuticles may be irregular, thickened, and distorted, and the lateral and palmar areas of the fingers may become rough and cracked, with irregular, "dirty" horizontal lines, resembling mechanic's hands • At times, the muscle strength appears normal, hence the term dermatomyositis sine myositis • DM usually occurs alone but may overlap with scleroderma and mixed connective tissue disease
DM often involves the hands as erythematous flat topped papules over the knuckles (Gottron’s sign) Periorbital violaceous erythema characterizes the classic heliotrope rash
Association with Malignancies • The relative risk of cancer is 2.4 for male and 3.4 for female patients, and a wide variety of cancers have been reportedusually gynaecological and carcinoid tumors
Inclusion Body Myositis • In patients 50 years of age, IBM is the most common of the inflammatory myopathies-often misdiagnosed as PM and is suspected only later when a patient with presumed PM does not respond to therapy • Weakness and atrophy of the distal muscles, especially foot extensors and deep finger flexors, occur in almost all cases of IBM- clue to early diagnosis • Falls due to quadriceps weakness, small muscle weakness • Weakness and accompanying atrophy can be asymmetric and selectively involve the quadriceps, iliopsoas, triceps, biceps, and finger flexors, resembling a lower motor neuron disease
Dysphagia is common, occurring in up to 60% • Sensory Normal • Disease progression is slow but steady, and most patients require an assistive device • 20% of cases-associated with systemic autoimmune or connective tissue diseases
Extra muscular Manifestations • Systemic symptoms, such as fever, malaise, weight loss, arthralgia, and Raynaud's phenomenon, especially when inflammatory myopathy is associated with a connective tissue disorder • Joint contractures, mostly in DM and especially in children • Dysphagia and gastrointestinal symptoms, due to involvement of oropharyngeal striated muscles and upper esophagus, especially in DM and IBM
Cardiac disturbances, including atrioventricular conduction defects, tachyarrhythmias, dilated cardiomyopathy, a low ejection fraction, and congestive heart failure, may rarely occur • Pulmonary dysfunction, due to weakness of the thoracic muscles, interstitial lung disease, or drug-induced pneumonitis-dyspnea, nonproductive cough, and aspiration pneumonia • Subcutaneous calcifications, in DM • Arthralgia's, synovitis, or deforming arthropathy with subluxation in the interphalangeal joints
Association with malignancies • In older age groups, the incidence of malignant conditions appears to be specifically increased only in patients with DM • The most common tumors associated with DM are ovarian cancer, breast cancer, melanoma, colon cancer, and non-Hodgkin lymphoma • Search that should be conducted for an occult neoplasm in adults with DM -are usually uncovered by abnormal findings in the medical history and physical examination
Complete annual physical examination with pelvic, breast, rectal examinations , urinalysis; complete blood count; blood chemistry tests; and a chest film • In Asians, nasopharyngeal cancer is common, and a careful examination of ears, nose, and throat is indicated • Whole-body PET scan should be considered
Autoantibodies and Immunogenetics • Autoantibodies against nuclear antigens (antinuclear antibodies) and cytoplasmic antigens are found in up to 20% of patients • antibody directed against the histidyl-transfer RNA synthetase, called anti-Jo-1, accounts for 75% of all the antisynthetases and is clinically useful -80% of patients with anti-Jo-1 antibodies have interstitial lung disease
Immune mechanism • In DM, humoral immune mechanisms are implicated, resulting in a microangiopathy and muscle ischemia • Necrosis of the endothelial cells, reduced numbers of endomysial capillaries, ischemia, and muscle-fiber destruction resembling microinfarctsoccur • By contrast, in PM and IBM a mechanism of T cell–mediated cytotoxicity is likely. • CD8 T cells, along with macrophages, initially surround and eventually invade and destroy healthy, nonnecrotic muscle fibers that aberrantly express class I MHC molecules.
DDs for similar complaints • polymyalgia rheumatica–no muscle pain • Arthritic joints- but no myositis • fibrositis and fibromyalgia–focal muscle diffuse in nature • Myopathy- Drug/Toxin • Neuro-muscular • Endocrine Disease • Infectious Myositis • Metabolic Storage Myopathies • Mitochondrial Myopathy
Criteria for Diagnosis of Inflammatory Myopathies • The clinically suspected diagnosis of PM, DM, or IBM is confirmed by analysis of serum muscle enzymes, EMG findings, and muscle biopsy
CK can be elevated as high as 50 fold, can be normal in some patients with DM or IBM • Serum glutamic oxaloacetic acid and glutamate pyruvate transaminases , LDH ,aldolase may be elevated. • MEG shows =myopathic potentials , short duration, low amplitude , polyphasic units • MRI may be used to guide the location • Muscle biopsy most sensitive and specific test for establishing diagnosis of inflammatory myopathy
Cross-sections of a muscle biopsy from a patient with inclusion body myositis demonstrate the typical features of vacuoles with lymphocytic infiltrates surrounding nonvacuolated or necrotic fibers (A), tiny endomysial deposits of amyloid visualized with crystal violet (B), cytochrome oxidase–negative fibers, indicative of mitochondrial dysfunction (C), and ubiquitous MHC-I expression at the periphery of all fibers (D).
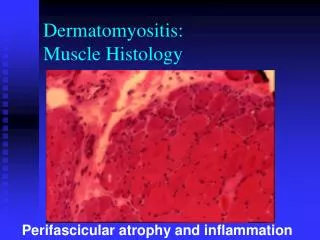
PM-Inflammation is primary – muscle fasciles • DM- endomysial inflammation –perivascular , interfasicularseptae and around the muscle than within • IBM – endomysial inflammation with T cell invasion. Nonvacoulated muscle fibres, basophilic granular deposits.
LABORATORY TESTS HIGH MUSCLE ENZYMES:- CPK ELEVATED ESR , CRP:- 50% POSITIVE ANA:- 50-80% AUTOANTIBODIES:- anti- RNP (MCTD) anti-PM/Scl (OVERLAP)
Myositis-specific AUTOANTIBODIES ANTI Jo-1 part of ANTI SYNTHETASE Ab’s Antibodies to the antigen- Aminoacyl-tRNAsynthetase, in 20-50% of PM>>DM ANTI SRP = anti signal recognition particle In 5% of PM ANTI Mi-2 in 10% of DM.
Treatment • Goal- Improve muscle strength, improve ADL, reduce – other manifestations like rash, dysphagia, dyspnea, fever. • CK Usually reduces with muscle strength improvement (but not conversely true) • Clinicians tend to chase CK inplace of muscle weakness • Need to discontinue immuno suppressants if no recovery or response
Glucocorticoids • Oral prednisone is initial choice – 1mg/kg/day initiated early -. 3-4 weeks later – taper -.10 weeks – 1mg/kg every day – reach lowest possible dose – till disease control. • Titer – effective muscle strength • > 3 months no response after high dose-unresponsive disease – next line immunosuppressive drug. • All DM /PM respond to some extent to prednisone
Other immunosuppressive drugs • 75% require additional drugs • Azathioprine –side effects less, effects for long term- dose 3mg/kg/daily • Methotrexate- faster onset of action, start dose 7.5mg/week x 3 weeks increase gradually 2.5mg/week to max 25mg/week. • Watch for pneumonitis – as compared to interstitial lung disease
Mycophenolate mofetil –faster action than azathioprine- 2.5mg/3gm/day BID doses well tolerated for long term use • Monoclonal anti CD20 antibody (rituximab) – shown response in certain trials • Cyclosporine- inconsistent benefits • Cyclophosphamide – 0.5-1g/sqmt – limited success. • Tacrolimus- some success
Immunomodulation • Ivlg improved not only strength and rash but also the underlying immunopathology. • The benefit is often short-lived(三8 weeks),repeated infusions every 6-8 weeks are generallyrequired to maintain improvement. • A dose of 2 g/kg divided over2-5 days per course is recommended . • Uncontrolled observations suggest that IVlg may a l s o be beneficial for patients with PM • Neither plasmapheresis nor leukapheresis appears to be effective in PM and DM.
sequential empirical approach • Step1 – High dose prednisone • Step 2- azathioprine , mycophenolate or mtx • Step3 – IV Ig • Step 4 –trial rituximab, cyclosporine , cyclophosphamide or tacrolimus
Prognosis • 5-year survival rate for treated patients with PM and DM is 95% and the 10-year survival rate is 84% • death is usually due to pulmonary, cardiac, or other systemic complications • Prognosis is severe for initial treatment delay • Older, Ca Pts have worserprognosis • DM responds more favourably than PM , IBM has least favourable prognosis • PM/DM most pts will recover full functional recovery -> maintenance therapy.