Download
1 / 99
990 likes | 1.63k Views
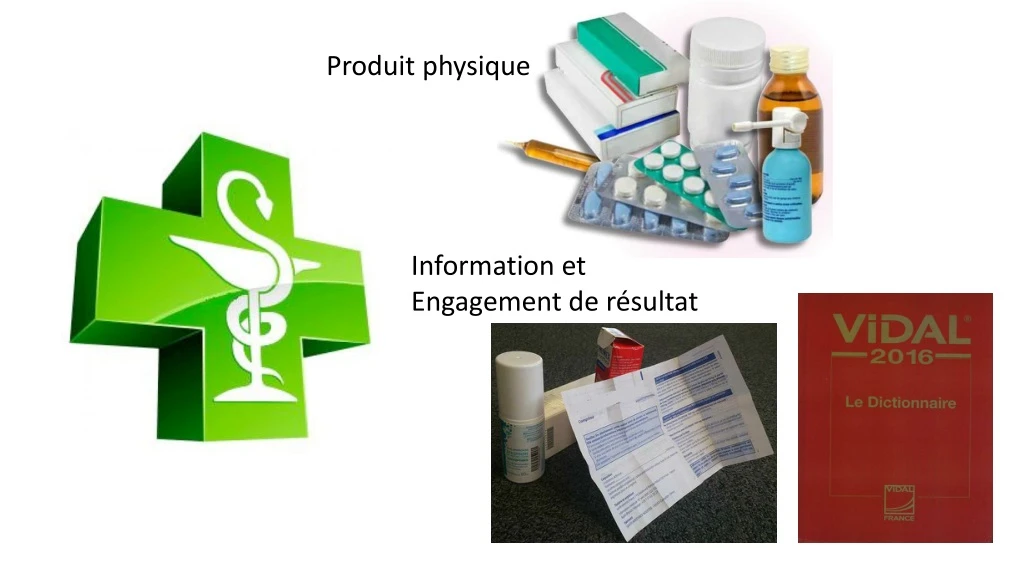
Produit physique. Information et Engagement de résultat. 1/Un « produit physique». -A-Substance active. B-Formulation galénique. -C-Emballage. 2/ Une « documentation ». établie au cours des différentes phases d’essais cliniques elle permet d’obtenir l’autorisation de mise sur le marché

E N D
Produit physique Information et Engagement de résultat
1/Un « produit physique» -A-Substance active • B-Formulation galénique -C-Emballage
2/ Une « documentation » établie au cours des différentes phases d’essais cliniques • elle permet d’obtenir l’autorisation de mise sur le marché • elle sert de base à l’information du prescripteur • ellesert à démontrer aux organismespayeurs le service médicalrendu et son amélioration.
Un médicament est un « produit » -1-Substance active • 2-Formulation galénique -3-Emballage
Drug substance (Active pharmaceutical ingredient) API Drug product (Dosage form; Finished product)
La Substance active • Substance ayant un effet pharmacologique susceptible de se traduire par une application thérapeutique • Substance pouvant être produite sous forme pure à grande échelle, de manière reproductible et économiquement et écologiquement viable • Substance susceptible d’être dosée ainsi que ses impuretés, ses produits de dégradation et ses métabolites • Toxique potentiel
Une substance ayant un effet pharmacologique susceptible de se traduire par une application thérapeutique
Le venin de serpent contient des substances ayant de puissants effets pharmacologiques AnCrod/ Virpinex est une protéase à sérine préparée à partir du venin d’une vipère qui permet de lyser les caillots formés lors d’un accident cérébrovasculaire
Une substance ayant un effet pharmacologique susceptible de se traduire par une application thérapeutique a. Dans le prolongement des essais pharmacologiques de discovery, il faudra montrer l’efficacité du principe actif dans des modèles animaux de la pathologie ciblée
Modèle animal d’obésité Modèle animal de migraine
Une substance ayant un effet pharmacologique susceptible de se traduire par une application thérapeutique a. Dans le prolongement des essais pharmacologiques de discovery, il faudra montrer l’efficacité du principe actif dans des modèles animaux de la pathologie ciblée b. Pour la mise en place des essais cliniques il faudra générer une relation dose-réponse entre la quantité administrée et l’effet pharmacologique et déterminer une durée d’action.
relation dose-réponse entre la quantité administrée et l’effet pharmacologique
2. Une substance pouvant être produite • pure, de qualité constante et de stabilité suffisante. • de manière reproductible (y compris les impuretés). La voie de synthèse sera souvent très différente de celle suivie lors de la découverte. • à grande échelle, de manière reproductible et économiquement et écologiquement viable
En 1995, apparait sur le marché le Saquinavir, le premier inhibiteur de la protéase du virus du sida Il est suivi en 1996, par un second médicament de la même classe thérapeutique: le Ritonavir (Norvir) qui est commercialisé sous la forme d’une solution buvable dosée à 80 mg/mL Les résultats sont spectaculaires
En 1998, une nouvelle forme cristalline très peu soluble commença à précipiter dans les flacons de Ritonavir (Norvir), menaçant d’entrainer un arrêt de la production. 20 mg/mL 170 mg/mL
3. Une substance susceptible d’être dosée au moyen de méthodes analytiques qui: • Devront permettre de doser le principe actif • - dans les fluides physiologiques • - dans les formulations • Devront faire l’objet d’une validation. • Devront aussi permettre le dosage • - des impuretés produites lors de la fabrication et du vieillissement • - des métabolites détectés lors des études de pharmacocinétique.
Métabolisme Hépatique du Ritonavir Structure des Onze Principaux Métabolites
Métabolisme Hépatique du Ritonavir identification des Onze Principaux Métabolites par Chromatographie
4. Un principe actif est un toxique potentiel qu’il faudra évaluer: • Tests cellulaires de mutagénicité et de génotoxicité. • Toxicité aigue après une dose unique ou sur une période de 24 heures • Toxicité sub-aigue selon un régime qui reflète l’application clinique envisagée. Réalisée sur deux espèces dont un non-rongeur. • Toxicité chronique, sur une période dépassant 6 mois permet d’évaluer les risques associés à l’usage prolongé de la substance. • Carcinogénicité : généralement sur des rongeurs pendant 2 ans. • Incidence sur la fertilité, tératogénicité, développement post-natal
Marge Thérapeutique relation dose-réponse entre la quantité administrée et l’effet pharmacologique
LaForme Galénique • 1. C’est la forme sous laquelle sera administré la substance active (comprimé, gélule, solution injectable, aérosol ….) • 2. Des formes simplifiées seront utilisées au cours des premiers essais cliniques (ex. des gélules remplies manuellement). • Elles doivent être stable pendant la durée des essais. • 3. La forme définitive sera développée durant les essais de phase I et II et adoptée lors des essais de phase III.
2/ Une « documentation » Etablie au cours des différentes phases d’essais cliniques • elle permet d’obtenir l’autorisation de mise sur le marché • elle sert de base à l’information du prescripteur et du patient • ellesert à démontrer aux organismespayeurs le service médicalrendu et son amélioration.
La documentation comprend les informations suivantes - Pharmacologie clinique • Indication(s) thérapeutiques - Contre-indications - Précautions - Effets secondaires - Dosage et administration - Interactions Médicamenteuses ….
Fabrication du Produit physique Information et Engagement de résultat
Cost of Good Sold / Sales Generics Brand Name Biologics • J Pharm Innov (2008) 3:30–40
Brand Name ~ 27% Biologics ~ 13% Fabrication du Produit physique Brand Name ~ 73% Biologics ~ 87% Information et Engagement de résultat
Link disease and target. Hit generation: HTS rational design In silico screen Hits confirmation Potency Cytotoxicity Preliminary animal efficacy Initial SAR Potency Selectivity PK/ ADME Toxproperties Full SAR Pharmacological profile Administration route Preclinicaltox D/D Interactions 20-100 people Safety 100-500 people Efficacy Dose ranging Safety 1000-10000 people Efficiency Safety Les “informations” sontacquises aux cours des phases précliniques et cliniques
Costs to discover and develop a new molecular entity p(TS): probability of technical success WIP: work in process NATURE Drug Discovery vol 9 | March 2010 | 203
Costs to discover and develop a new molecular entity NATURE Drug Discovery vol 9 | March 2010 | 203
R&D cost + Cost of Production + MG&A For one Treatment Treatment Cost per patient = Number of patients 1000 millions 10.000.000 patients Blockbuster Model = 100 $ 10.000.000 400 millions $ Orphan Drug Model 10.000 patients = 40.000 $ 10.000
The Shift to High-Priced Innovator Drugs in the USA http://www.evaluategroup.com/public/EvaluatePharma-Free-Access.aspx
The Shift to High-Priced Innovator Drugs in the USA http://www.evaluategroup.com/public/EvaluatePharma-Free-Access.aspx
Clinical Development of a new molecular entity NATURE Drug Discovery vol 9 | March 2010 | 203
the drug development process is divided into six distinct phases: • Regulatory preclinical studies • Phase-Ia • Phase-Ib • Phase-IIa • Phase-IIb • Phase-III
The preclinical phase : • The preclinical phase is defined as the phase • from the first good laboratory practice (GLP) toxicology dose (at least two mammalian species), prior to human trials authorization through • to an investigational new drug (IND, US) application or first clinical trial application (CTA, EU) before first-in-human (FIH) testing.
Phase-I • Phase I is the phase that includes the First-In-Human (FIH) trials within a small trial population (typically <50 patients). • The tested range of doses will usually be a fraction of the dose (1/100) that caused harm in animal tests. • Comprises safety, tolerability and pharmacokinetics/dose ranging studies. • These studies are often conducted in healthy volunteers, but in some indications (for example, oncology) they can include patients. • Phase I trials are usually conducted in a clinical trial clinic, where the subject can be observed by full-time • staff. These clinical trial clinics are often run by CROs who conduct these studies on behalf of pharmaceutical companies.
Phase I trials are subdivided into: • Phase Ia: • In single ascending dose studies (SAD), small groups of subjects (3-5) are given a single dose of the drug. • If they do not exhibit any adverse side effects, and the pharmacokinetic data are in line with predicted safe values, the dose is escalated (X2), and a new group of subjects is then given a higher dose. • This is continued until pre-calculated pharmacokinetic safety levels are reached, or intolerable side effects start showing up (the drug is said to have reached the maximum tolerated dose (MTD).
TGN1412, a monoclonal antibody is a TCR-independent agonist binding to CD28 present at the surface of CD4+ T-cells. The phase-I trial conducted by Parexelwas a double-blind, randomized, placebo-controlled study, with two of the eight subjects receiving a placebo, and six receiving 0.1 mg per kg (1/500th of the highest dose found safe in preclinical experiments with macaques). Within half an hour all six subjects experienced a catastrophic systemic organ failure corresponding to a cytokine release syndrome resulting in angioedema, swelling of skin and mucous membranes.
The therapeutic Index is a comparison of the amount of a therapeutic agent that causes the therapeutic effect to the amount that causes toxicity. A phase-I trial is not designed to evaluate the therapeuticeffect
Phase I trials are subdivided into: • Phase Ib: • In multiple ascending doses studies (MAD), a group of subjects receives multiple low doses of the drug, while samples (of blood, urine …) are collected at various time points and analyzed to acquire information on how the drug is processed within the body. • The dose is subsequently escalated. • Dose levels and dosing frequency are chosen in order to achieve steady state therapeutic drug levels.
Phase-II • Phase II trials are aimed at evaluating the candidate drug’s efficacy in a patient population, leading up to clinical proof of concept (PoC). • Phase II trials are also subdivided into Phase IIa and Phase IIb: • Phase IIa studies are generally smaller (typically <200 patients) and designed to mainly address early evidence of drug activity and to assess dosing requirements. • Phase IIb studies include larger numbers of patients (typically <400 patients) and are designed to demonstrate clinical proof of concept and an understanding of dose response. • Genetic testing for polymorphic metabolism enzymes (Cyp2D6 or 2C9…), are performed particularly when there is evidence of variation in metabolic rate between individuals.
Phase II clinical trials of ADX10059 (mGluR5 allostericinhibitor) in Gastro-Esophageal Reflux Disease (GERD) Phase IIatrial: Two days single-blind study in 24 patients. Objectives: evaluate the effect of a single dose using continuous 24-hour pH recording in the lower esophagus, and on the occurrence of patient-recorded clinical symptoms of GERD. Phase IIbtrial: Two weeks of administration twice daily in 103 GERD patients. Objectives: evaluate the effects on esophageal function and reflux events using impedance pH monitoring and esophageal manometry.
Phase-III • Phase III trials are designed to assess the effectiveness of the new drug and its value in clinical practice. • Phase III studies are randomized trials involving large patient groups (300–3,000 or more depending upon the disease and the end points). Due to the difficulty in recruiting patients, they are in general multicenter trials. • Phase-III represent the definitive assessment of how effective the drug is, in comparison with the current reference treatment (or a placebo if no approved reference treatment is available). • This assessment is based on the achievement of predetermined end-points: • “Results, condition or events associated with individual study patients that are used to assess study treatments”. (FDA) • Primary end point: single endpoint parameter that will define the success or the failure of the drug. • Secondary end point(s): other endpoints pre-specified, may be powered for hypothesis testing
Direct Endpoints (FDA) All drugs have safety risks. Therefore, the only reason that a patient would want to take a drug would be if the drug: – improved survival – resulted in a benefit that was detectable by the patient (improvement in symptoms, in functional capacity), or – decreased the chances of developing a condition or disease complication that is itself apparent to the patient and is undesirable (e.g. stroke). Therefore, a primary endpoint should be a direct measure of one of these. A primary endpoint should generally not be a measure of something that is not important to the patient. (exception: validated surrogate endpoint).
Surrogate endpoint – used instead of direct endpoint – Ideally, the surrogate parameter should exist within the therapeutic pathway between the drug and meaningful benefit – i.e. the drug results in the therapeutic benefit by virtue of its effect on the surrogate parameter. Changes induced by a therapy on a surrogate endpoint are expected to reflect changes in a clinically meaningful endpoint ex hepatitis C: SVR-24 Sustained Viral Response (Detection of HCV RNA negative 24 weeksafter the end of treatment).
Hepatitis C Natural History The full evolution of the diseasetakes 20 to 40 years itis impossible to evaluate the success of a therapy on direct clinical end-points. http://www.sciencedirect.com.doc-distant.univ-lille2.fr/science/article/pii/S1521691812000923
Goal of hepatitis C treatment : Primary end-point SVR 24: Detection of HCV RNA negative 24 weeksafter 48 weeks of treatment.








![[Nom du produit]](https://cdn2.slideserve.com/4968904/nom-du-produit-dt.jpg)