Download

1 / 119
1.21k likes | 1.63k Views
Amino acid metabolism. The first of them told him so, with the customary prison sign of Death—a raised finger—and they all added in words, “Long live the Republic!” A Tale of Two Cities. Charles Dickens . Functions of Amino Acid Metabolism Proteins constantly undergo breakdown and synthesis

E N D
Amino acid metabolism The first of them told him so, with the customary prison sign of Death—a raised finger—and they all added in words, “Long live the Republic!” A Tale of Two Cities. Charles Dickens
Functions of Amino Acid Metabolism • Proteins constantly undergo breakdown and synthesis • Total protein turnover in a well-fed, adult human is estimated at about 300 g/day, of which approximately 100 g is myofibrillar protein, 30 g is digestive enzymes, 20 g is small intestinal cell protein, and 15 g is hemoglobin • The remainder is accounted for by turnover of cellular proteins of various other cells (e.g., hepatocytes, leukocytes, platelets) and a very small amount is lost as free amino acids in urine • Protein turnover is not completely efficient in the reutilization of amino acids. Some are lost by oxidative catabolism, while others are used in synthesis of non-protein metabolites • For this reason, a dietary source of protein is needed to maintain adequate synthesis of protein • There is no distinct storage form for amino acids in the body • The turnover of some proteins, particularly those in muscle, is increased under conditions of fasting and starvation
Plants and some bacteria synthesize all 20 amino acids. Humans (and other animals) can synthesize some (the non-essential amino acids) but require the others to be supplied by the diet (the essential amino acids) • The eight essential amino acids are isoleucine, leucine, lysine, methionine, phenylalanine,threonine, tryptophan, and valine • Under certain conditions, some nonessential amino acids may become essential –these amino acids are known as conditionally essential amino acids • Although arginine and histidine are not essential amino acids in adults, their rates of synthesis in neonates are not adequate to meet their requirements for optimal growth; they should, therefore, be supplied in the diet • Synthesis of cysteine and tyrosine is dependent on adequate dietary intake of methionine and phenylalanine, respectively
Glutamine, a nitrogen donor in the synthesis of purines and pyrimidines required for nucleic acid synthesis, aids in growth, repair of tissues, and promotion of immune function • Enrichment of glutamine in nutrition augments recovery of seriously ill patients • Exogenous arginine also becomes essential in cases of sepsis (the presence of various pathogenic organisms, or their toxins, in the blood or tissues), when there is both a decrease in endogenous synthesis of arginine and an increased requirement of arginine for the synthesis of protein and nitric oxide • For protein synthesis to occur, all amino acids must be present in sufficient quantities. Absence of any one essential amino acid leads to cessation of protein synthesis, catabolism of unused amino acids, increased loss of nitrogen in urine, reduced growth • Negative nitrogen balance exists when the amount of nitrogen lost from the body (as nitrogen metabolites excreted in urine and feces) exceeds that taken in
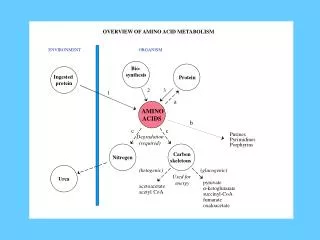
Negative nitrogen balance occurs in malabsorption syndromes, fever, trauma, cancer, and excessive production of catabolic hormones • When the dietary nitrogen intake equals nitrogen losses, the body is in nitrogen balance. In normal adults, anabolism equals catabolism • When nitrogen intake exceeds nitrogen losses, there is a positive nitrogen balance, with anabolism exceeding catabolism. In this case, the body retains nitrogen as tissue protein, which is a characteristic of active growth and tissue repair • So, in general, there are three major fates for amino acids: • Synthesis of new proteins for growth or repair • Synthesis of a range of nitrogen-containing small compounds • Catabolism. This results, eventually, in formation of ammonia and small carbon-containing compounds. The carbon skeletons are used for the synthesis of glucose and triacylglycerol or for energy production. The ammonia is converted to urea
Protein Digestion • The digestion of proteins begins in the stomach and is completed in the intestine • The enzymes that digest proteins are produced as inactive precursors (zymogens) • The inactive zymogens are secreted from the cells in which they are synthesized and enter the lumen of the digestive tract, where they are cleaved to smaller forms that have proteolytic activity • These active enzymes have different specificities; no single enzyme can completely digest a protein • However, by acting in concert, they can digest dietary proteins to amino acids and small peptides, which are cleaved by peptidases associated with intestinal epithelial cells • The acid in the stomach lumen alters the conformation of pepsinogen so that it can cleave itself, producing the active protease pepsin
Thus, the activation of pepsinogen is autocatalytic • Dietary proteins are denatured by the acid in the stomach. This serves to inactivate the proteins and partially unfolds them such that they are better substrates for proteases • However, at the low pH of the stomach, pepsin is not denatured and acts as an endopeptidase, cleaving peptide bonds at various points within the protein chain • Although pepsin has a fairly broad specificity, it tends to cleave peptide bonds in which the carboxyl group is provided by an aromatic or acidic amino acid • A proteolytic enzyme secreted by gastric mucosa of infants is chymosin (rennin), which functions to clot milk and promote its digestion by preventing rapid passage from the stomach • Chymosin hydrolyzes casein, a mixture of several related milk proteins, to paracasein, which reacts with Ca 2+ to yield the insoluble curd • As the gastric contents empty into the intestine, they encounter the secretions from the exocrine pancreas
One of these secretions is bicarbonate, which, in addition to neutralizing the stomach acid, raises the pH such that the pancreatic proteases, which are also present in pancreatic secretions, can be active • These pancreatic proteases are also secreted as zymogens. Because the active forms of these enzymes can digest each other, it is important for their zymogen forms all to be activated within a short span of time • Trypsin, elastase, and chymotrypsin are endopeptidases. Carboxypeptidases are exopeptidases • The combined action of these enzymes produces oligopeptides having two to six amino acid residues and free amino acids • Exopeptidases produced by intestinal epithelial cells act within the brush border and also within the cell • Aminopeptidases, located on the brush border, cleave one amino acid at a time from the amino end of peptides. Intra-cellular peptidases act on small peptides that are absorbed
Protein Digestion The Specificity of Proteases
Absorption of Amino Acids • Amino acids are absorbed from the lumen of the small intestine principally by semi-specific Na+-dependent transport proteins in the luminal membrane of the intestinal cell brush border, similar to that already seen for carbohydrate transport • At least six different Na+-dependent amino acid carriers are located in the apical brush border membrane of the epithelial cells • These carriers have an overlapping specificity for different amino acids (for neutral amino acids, proline and hydroxyproline, …) • As with glucose transport, the Na+-dependent carriers of the apical membrane of the intestinal epithelial cells are also present in the renal epithelium • The amino acids are then transported out of the cell into the portal circulation principally by facilitated transporters in the serosal membrane
Di- and tripeptides enter the epithelial cells through symport with H+. The H+ gradient is maintained by the Na+ - H+ exchanger • Amino acids that enter the blood are transported across cell membranes of the various tissues principally by Na+-dependent cotransporters and, to a lesser extent, by facilitated transporters • In this respect, amino acid transport differs from glucose transport, which is Na+-dependent transport in the intestinal and renal epithelium but facilitated transport in other cell types
Disorders of Protein Malnutrition, Digestion and Amino Acid Absorption • The principal causes of protein maldigestion and malabsorption are diseases of the exocrine pancreas (such as cystic fibrosis) and small intestine • Defects in neutral amino acid transport (Hartnup disease), in basic amino acids and cystine (cystinuria),… have been reported • The clinical severity of these disorders is usually minimal and relates to the loss of amino acids or relative insolubility of certain amino acids in the urine • Kwashiorkor (in Ga, "the disease the first child gets when the second is on the way") is a form of protein-calorie malnutrition that is caused by dietary protein deficiency and is often exacerbated by infection • The classic presentation, particularly in poorer countries, is a young child who has been weaned to an adult diet that lacks sufficient protein to sustain healthy growth
The characteristics of kwashiorkor include growth failure, edema, fatty liver, and “flaky paint” patches of skin • Because of the low protein intake, there is a deficiency of amino acids for synthesis of serum albumin and other plasma proteins, resulting in edema and the characteristic swollen abdomen and limbs • The situation is made worse by the availability of ample dietary carbohydrates, which stimulate insulin secretion and thus inhibit mobilization of amino acids from skeletal muscle • This dietary carbohydrate also provides substrate for fatty acid synthesis, which in the absence of adequate protein synthesis results in fatty liver and hepatomegaly • The clinical manifestation of a diet deficient in both protein and energy is marasmus, (from the Greek "to waste away") which results in severe muscle wasting and marked growth retardation
Protein Turnover • The amino acid pool within cells is generated both from dietary amino acids and from the degradation of existing proteins within the cell • All proteins within cells have a half-life (t1/2), a time at which 50% of the protein that was synthesized at a particular time will have been degraded • The rates of protein turnover vary enormously, depending on the nature of the protein, the condition of the subject and the tissue • Proteins (mainly enzymes) in the liver are replaced every few hours or days whereas structural proteins (e.g. collagen, contractile proteins) are stable for several months • Why should turnover occur? • It helps remove defective proteins and replace them with normal ones • The regulation of hormone and enzyme levels
There are two main pathways for protein degradation: the lysosomal-autophagic system and the ubiquitin-proteasome degradation pathway • The quantitative importance of each pathway varies from one tissue to another and from one protein to another • Although hydrolysis of the peptide bonds does not involve ATP, the various processes of protein degradation require considerable expenditure of energy, possibly more than is required for protein synthesis • Protein turnover contributes at least 20% to resting energy expenditure (basal metabolic rate) • The Lysosomal –Autophagic System • In general, extracellular, membrane-associated, and long-lived intracellular proteins are degraded in lysosomes by ATP-independent processes
A variety of proteases known as cathepsins and peptidases exist so that proteins can be hydrolysed completely to amino acids • The pH within the lysosome is 4.5–5.0 and all lysosomal enzymes exhibit low pH optima • This ensures that, if they leak into the cytosol, their activity is very low and little damage is done • This low pH within the organelle is maintained by a proton pump, driven by the hydrolysis of ATP, thus contributing to the energy • Proteins enter the lysosome by two main mechanisms: • Vesicles transport extracellular particles and membrane proteins into the cell, where they fuse with the lysosomes (endocytosis) • The endoplasmic reticulum engulfs some cytosolic proteins to form vesicles which fuse with the lysosomes
The Ubiquitin-Proteasome System • In general, the degradation of regulatory proteins with short half-lives and of abnormal proteins occurs in the cytosol, through the ubiquitin-proteasome system • This system is quantitatively the most important process for protein breakdown in mammalian cells. It is so named because it involves the proteolytic enzyme (the proteasome), and the protein ubiquitin • The proteasome is a very large complex of at least 50 subunits. It is present in a wide variety of tissues and can constitute up to 1% of soluble protein in a cell • The catalysis occurs within the central core of the molecule and ATP hydrolysis is required to ‘drive’ the protein into the core. • Before the complex can break down proteins, the protein must first be ‘tagged’ by complexing with ubiquitin • Ubiquitin, so named because it is present in all eukaryotic cells, is a small (8.5 kDa, 76 aa residues) protein
The protein substrate has amino groups in the side chains of its Lys residues • Ubiquitin has got a C-terminal Gly. The carboxyl group of this Gly forms an isopeptide bond with the amino group of Lys • Oftentimes, the target protein is polyubiquitinylated, in which additional ubiquitin molecules are added to previous ubiquitin molecules • The Lys on one ubiquitin molecule serve as internal acceptors for the carboxyl of Gly on another ubiquitin molecule, allowing the formation of a chain • The residue present at its amino terminal affects whether a protein is ubiquitinated. Amino terminal Met or Ser retards, whereas Asp or Arg accelerates ubiquitination • In addition, many proteins that contain regions rich in the amino acids proline (P), glutamate (E), serine (S), and threonine (T) have short half-lives. These regions are known as PEST sequences
Three enzymes are involved in the attachment of ubiquitin to a protein: E1 (an activating enzyme), E2 (a conjugating enzyme), and E3 (a ligase) • Some pathological conditions vividly illustrate the importance of the regulation of protein turnover. For example, human papilloma virus (HPV) encodes a protein that activates a specific E3 enzyme. The enzyme ubiquitinates the tumor suppressor p53 and other proteins that control DNA repair, which are then destroyed. The activation of this E3 enzyme is observed in more than 90% of cervical carcinomas
The proteasome degrades the targeted protein, releasing intact ubiquitin that can again mark other proteins for degradation • The basic proteasome is a cylindrical 20S protein complex with multiple internal proteolytic sites • ATP hydrolysis is used both to unfold the tagged protein and to push the protein into the core of the cylinder • Additional subunits, some of which catalyze ATP hydrolysis, form a cap which adds to one or both ends of a 20S proteasome to give a larger 26S proteasome. • Different cap complexes alter the specificity of the proteasome • For example, the PA700 cap is required for ubiquitinated proteins, whereas the PA28 cap targets only short peptides to the complex • After the target protein is degraded, the resultant amino acids join the intracellular pool of free amino acids
Some key intracellular processes, which involve proteasomal degradation include: • The cell cycle • The concentrations of specific proteins known as cyclins (that regulate the cell cycle) is regulated by synthesis and degradation • Transcription factors • These factors activate the expression of genes. In order to carry out their regulatory function, they must have short half lives. Their degradation is carried out by this system • Formation of antigens from the degradation of pathogens • The proteolytic system hydrolyses proteins of pathogens that are present within the host cell to produce a short peptide which forms a complex with a specific protein, known as the major histocompatibility complex (MHC) protein • The peptide is, in fact, the antigen
At the plasma membrane, the MHC protein locates within the membrane and the small peptide sits on the outside of the membrane, where it can interact with the receptor on T- lymphocytes • Defects in Protein Degradation and Diseases • The importance of the proteasomal-ubiquitin system in the degradation of cellular proteins or proteins of pathogens suggests that any defects in this system could result in disease • Prion diseases and amyloid diseases (such as Alzheimer’s and Parkinson’s) involve the aggregation of degradation-resistant proteins • Failure to control the rate of degradation of cyclins could lead to their over-expression, increasing the risk of tumour development • Infectious agents may hijack cell machinery involved in ubiquitination and protein degradation system
Interorgan Relation in Amino Acid Metabolism • In the fed state, amino acids released by digestion of dietary proteins (except the greater portion of branched-chain amino acids) travel through the hepatic portal vein to the liver, where they are used for the synthesis of proteins, particularly the blood proteins, such as serum albumin • Excess amino acids are converted to glucose or to triacylglycerols. The latter are then packaged and secreted in VLDL. The glucose produced from amino acids in the fed state is stored as glycogen or released into the blood if blood glucose levels are low • Amino acids that pass through the liver are converted to proteins in cells of other tissues • Muscle generates over half of the total body pool of free amino acids in the post-absorptive state, and liver is the site of the urea cycle enzymes necessary for disposal of excess nitrogen
Muscle and liver thus play major roles in maintaining circulating amino acid levels • Free amino acids, particularly alanine and glutamine, are released from muscle into the circulation. Alanine, which appears to be the vehicle of nitrogen transport in the plasma, is extracted primarily by the liver • Glutamine is extracted by the gut and the kidney (among other tissues), both of which convert a significant portion to alanine • The kidney provides a major source of serine for uptake by peripheral tissues, including liver and muscle • Branched-chain amino acids, particularly valine, are released by muscle and taken up predominantly by the brain • Metabolism of Ammonia • Transamination is the major process for removing nitrogen from amino acids. In most instances, the nitrogen is transferred as an amino group from the original amino acid to α- ketoglutarate, forming glutamate
Post-absorptive State Fed State
The original amino acid is converted to its corresponding α-keto acid • For example, the amino acid aspartate can be transaminated to form its corresponding α-keto acid, oxaloacetate • In the process, the amino group is transferred to α-keto-glutarate, which is converted to its corresponding amino acid, glutamate • All amino acids except lysine, threonine and proline undergo transamination reactions • The enzymes catalyzing these reactions are known as transaminasesor aminotransferases • Pyridoxal phosphate (PLP), is the cofactor in transamination reactions • The aldehyde group of PLP can accept the α-amino group from an amino acid, generating pyridoxamine phosphate, which in turn donates that amino group to an α-ketoacid, regenerating PLP
The Mechanism of Transamination Reactions
The activated intermediate in this process is a Schiff base • PLP-containing enzymes also catalyze many other reactions involving amino acids, including the decarboxylation reactions involved in the synthesis of the neurotransmitters; PLP is also involved in the glycogen phosphorylase reaction • Because transamination reactions are readily reversible, they can be used to remove nitrogen from amino acids or to transfer nitrogen to α-keto acids to form amino acids • Thus, they are involved both in amino acid degradation and in amino acid synthesis • Liver is the major site of aminotransferase activity • The two principal liver transaminases are alanineaminotransferase (ALT), which catalyzes the reaction alanine + α-ketoglutarate → pyruvate + glutamate and aspartate aminotransferase (AST), which catalyzes the reaction aspartate + α-ketoglutarate → oxaloacetate + glutamate
Most of the NH4+ generated when muscle proteins are broken down during a fast is exported in the form of alanine • In the liver, ALT catalyzes the transamination of alanine, generating pyruvate which can be utilized for gluconeogenesis, and glutamate which provides nitrogen atoms for urea synthesis • Some of the glutamate nitrogen is released as ammonium ions by the enzyme glutamate dehydrogenase • Concurrently, AST utilizes some of the glutamate to generate aspartate by transfer of the amino group from glutamate to oxaloacetate • The NH4+ from the glutamate dehydrogenase reaction and the aspartate from the AST reaction provide the two nitrogens for urea synthesis • Aminotransferases are intracellular enzymes that have both cytosolic and mitochondrial isoforms
When there is liver damage, as occurs with cirrhosis or viral hepatitis, these aminotransferase are released from the hepatocytes • Increased plasma levels of ALT and AST are thus markers of liver damage • In the older clinical literature, these enzymes are sometimes referred to as SGPT (serum glutamate:pyruvatetransaminase) and SGOT (serum glutamate:oxaloacetatetransaminase), respectively • Generation of Ammonium Ions from Amino Acids • Two steps are required to generate ammonium ions from most of the common amino acids • The first is the aminotransferase reaction and the second step is the NAD+/NADP+-dependent oxidative deaminationof glutamate, which releases a free ammonium ion, regenerating α-ketoglutarate in the process
This reaction, which occurs in the mitochondria of most cells, is readily reversible; it can incorporate ammonia into glutamate or release ammonia from glutamate • The nitrogen in glutamate can be given off for biosynthesis or the removal in the form of urea • In general, glutamate can be thought of as a reservoir for amino groups while alanine and glutamine are the major transport forms of nitrogen in the blood
In addition to glutamate, a number of amino acids release their nitrogen as NH4+ • Histidine may be directly deaminated to form NH4+ and urocanate • The deaminations of serine and threonine are dehydration reactions that require pyridoxal phosphate and are catalyzed by serine and threoninedehydratases, respectively • Serine forms pyruvate, and threonine forms α-ketobutyrate. In both cases, NH4+ is released • Glutamine and asparagine contain R group amides that may be released as NH4+ by deamidation • Asparagine is deamidated by asparaginase, yielding aspartate and NH4+ . Glutaminase acts on glutamine, forming glutamate and NH4+ • The glutaminase reaction is particularly important in the kidney, where the ammonium ion produced is excreted directly into the urine as a buffer
Ammonium ion is also generated through the deamination of AMP to IMP (inosinemonophosphate) by adenosine monophosphatedeaminase • This reaction is especially active in exercising muscle which generates AMP. When ATP levels are low, muscle can generate additional ATP directly from ADP by means of the (adenylate kinase reaction • Removal of the resulting AMP is necessary if the reaction is to continue • The pathway by which AMP is deaminated to IMP and IMP is subsequently utilized for resynthesis of AMP is referred to as the purine nucleotide cycle • This cycle is also active in the brain but not in the liver • D-amino acids from bacterial cell walls and cooked food are metabolized by D-amino acid oxidasethat is active in the liver and the kidneys; the products are a keto acid, FADH2 and NH4+
Bacteria in the gut also act on amino acids, urea and other nitrogen containing molecules to release free ammonia Sources of Free Ammonia
The Urea Cycle • Humans excrete excess dietary nitrogen primarily as urea in the urine • At plasma concentrations greater than 50 μM, ammonia (at physiological pH, 98.5% exists as NH4+ ) is toxic to the CNS • Different animals excrete excess nitrogen as ammonia, uric acid, or urea • The aqueous environment of fish, which are ammonotelic(excrete ammonia), compels them to excrete water continuously to facilitate excretion of the highly toxic molecule ammonia • Birds, which must conserve water and maintain low weight, are uricotelicand excrete uric acid as semisolid guano • Many land animals, including humans, are ureotelic and excrete nontoxic, water-soluble urea • The urine of humans contains nitrogenous compounds other than urea, including uric acid, creatinine, and ammonia
These molecules serve other, distinct functions or represent breakdown products of certain metabolites • For example, creatinine is a breakdown product of muscle creatine phosphate and, as such, provides the clinician with a convenient measure of muscle mass • Uric acid is the end product of purine catabolism • The kidney also excretes some nitrogen directly in the form of ammonium ions, which serve to buffer acidic, anionic waste products such as β-hydroxybutyrate, acetoacetate, and sulfate • Excretion of ammonium ions is thus increased during ketoacidosis and other metabolic conditions where excess organic acids are produced • The Reactions of the Urea Cycle • Synthesis of Carbamoyl Phosphate • In the first step of the urea cycle, NH4+ , bicarbonate, and ATP react to form carbamoyl phosphate. The cleavage of 2 ATPs is required to form the high-energy phosphate bond of CP
The enzyme that catalyzes this reaction is carbamoyl phosphate synthetase I (CPS I), and it is localized to mitochondria • Carbamoyl phosphate synthetase II (CPS II) is a cytosolic enzyme that generates carbamoyl phosphate for pyrimidine synthesis. The nitrogen donor in this case is glutamine • The Synthesis of Citrulline • Orinthinetranscarbamoylasecatalyzes the transfer of the carbamoyl group from carbamoyl phosphate to the amino group in the side chain of the amino acid ornithine, generating citrulline. The high- energy phosphate bond of carbamoyl phosphate provides the energy required for this reaction, which occurs in mitochondria
Citrulline is transported out of the mitochondrion in exchange for ornithine (antiport) • Formation of Argininosuccinate • In the cytosol, citrulline reacts with aspartate, the second source of nitrogen for urea synthesis, to produce argininosuccinate • This reaction, catalyzed by argininosuccinate synthetase, is driven by the hydrolysis of ATP to AMP and PPi • The reaction is driven forward by hydrolysis of pyrophosphate to inorganic phosphate
Formation of Arginine and Fumarate • Argininosuccinate is cleaved by argininosuccinate lyaseto form fumarate and arginine • Fumarate is produced from the carbons of argininosuccinate provided by aspartate • Fumarate is converted to malate (using cytoplasmic fumarase), which is used either for the synthesis of glucose by the gluconeogenic pathway or for the regeneration of oxaloacetate by cytosolic malate dehydrogenase
The oxaloacetate that is formed is transaminated to generate the aspartate that carries nitrogen into the urea cycle • Thus, the carbons of fumarate can be recycled to aspartate The Krebs Bicycle
When these microorganisms die, their proteins are digested, releasing the amino acids to be absorbed into the blood • Some of the ammonia produced by urease travels to the liver and is converted back to urea • Ornithine generated by arginase is transported back into mitochondria to continue the cyclic process of urea synthesis • The urea cycle in effect changes ornithine to arginine Urea Salvage
Some of the enzymes of the urea cycle are present in tissues other than the liver • The intestinal mucosa can convert ornithine to citrulline, and the kidney can convert the resultant citrulline to arginine • However, since the kidney and intestine lack arginase, they cannot synthesize urea • Regulation of Urea Synthesis • In general, the urea cycle is regulated by substrate availability: the higher the rate of ammonia production, the higher the rate of urea formation • Regulation by substrate availability is a general characteristic of disposal pathways, such as the urea cycle, which remove toxic compounds from the body • Two other types of regulation control the urea cycle: allosteric activation of CPSI by N-acetylglutamate (NAG) and induction/repression of the synthesis of urea cycle enzymes
NAG is formed specifically to activate CPSI; it has no other known function in mammals (just like F 2,6-BP) • The synthesis of NAG from acetyl-CoA and glutamate is stimulated by arginine • Thus, as arginine levels increase within the liver, two important reactions are stimulated • The first is the synthesis of NAG, which will increase the rate at which carbamoyl phosphate is produced • The second is to produce more ornithine (via the arginase reaction), such that the cycle can operate more rapidly • The induction of urea cycle enzymes occurs in response to conditions that require increased protein metabolism, such as a high-protein diet or prolonged fasting • In both of these physiologic states, as amino acid carbon is converted to glucose, amino acid nitrogen is converted to urea • Stress (like in the case of sepsis, burns or trauma) also induces urea cycle enzymes
Hyperammonemia and Treatment • The finding of elevated blood levels of ammonia is evidence that the conversion of ammonia to urea is impaired in some way • Hyperammonemia in adults is usually the consequence of impaired liver function, secondary to liver disease (e.g., cirrhosis), organ transplantation, or chemotherapy • Transient hyperammonemia is often seen in premature neonates with immature liver function and/or inadequate hepatic blood flow
Impaired urea synthesis may also be the result of a genetic defect in one of the enzymes of the urea cycle • Regardless of its etiology, hyperammonemia is usually accompanied by increased plasma levels of glutamine, the amino acid that the brain uses as a vehicle to export excess ammonium ions • Ammonia is toxic to the central nervous system, where it can cause both acute encephalopathy and long-term irreversible brain damage; however, the pathophysiologic mechanisms are not fully understood • One possible cause is the increased synthesis of the neurotransmitters glutamate and GABA and subsequent derangements of neurotransmission • Another possible mechanism for ammonia toxicity in the brain involves the depletion of TCA-cycle intermediates by diversion of α-ketoglutarate to glutamate and glutamine synthesis, which would compromise the ability of the neural cells to generate ATP
Treatment for hyperammonemia involves dialysis to remove the excess ammonia • In acute cases, oral sodium benzoate and sodium phenylbutyrateare sometimes administered to provide alternate pathways for nitrogen excretion as hippurate and phenylacetylglutamine, respectively • The body then has to use its nitrogen to resynthesize the excreted amino acid
Protein intake should also be severely restricted in patients with hyperammonemia • At the same time, it is important to provide adequate intake of carbohydrates to minimize further catabolism of endogenous protein • Neonatal hyperammonemiais due to inborn errors of urea-cycle enzymes • In these defects, low blood urea nitrogen (BUN) accompanies the hyperammonemia • The most common inborn error of the urea cycle is a deficiency of ornithinetranscarbamoylase, an X-linked disorder • Ammonia intoxication is most severe when the metabolic block occurs at CPS I or OTC, because if citrulline can be synthesized, some ammonia has already been removed by being covalently linked to an organic metabolite • If the enzyme defect is in argininosuccinate lyase, massive arginine supplementation is beneficial