Download
1 / 39
390 likes | 602 Views
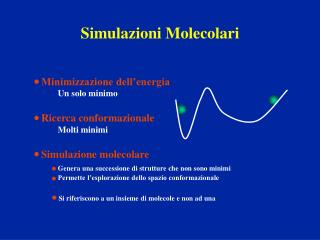
Minimizzazione dell’energia Un solo minimo Ricerca conformazionale Molti minimi Simulazione molecolare Genera una successione di strutture che non sono minimi Permette l’esplorazione dello spazio conformazionale Si riferiscono a un insieme di molecole e non ad una.

E N D
Minimizzazione dell’energia Un solo minimo Ricerca conformazionale Molti minimi Simulazione molecolare Genera una successione di strutture che non sono minimi Permette l’esplorazione dello spazio conformazionale Si riferiscono a un insieme di molecole e non ad una Simulazioni Molecolari
Calcolo delle proprietà di un sistema Proprietà energetiche Proprietà strutturali Proprietà termodinamiche Proprietà dinamiche Ricerca conformazionale Studio del moto delle molecole Applicazioni macroscopico
I metodi di simulazione molecolare permettono di studiare e prevedere il comportamento di sistemi macroscopici costituiti da un numero enorme di molecole ( 1023) Ciò è ottenuto tramite l’uso di modelli ridotti del sistema macroscopico che contengono un numero ridotto di molecole (10-1000) Le tecniche di simulazione molecolare sono due: Monte Carlo Dinamica molecolare Generalità
In generale il valore di una proprietà di un sistema dipende dalla posizione e dai momenti pi=mivi delle N particelle che variano nel tempo x1(t), y1(t), z1(t), x2(t), y2(t) , z2(t) ……., xN(t), yN(t), zN(t) px1(t), py1(t), pz1(t), px2(t), py2(t) , pz2(t) ……., pxN(t), pyN(t), pzN(t) Il valore istantaneo della proprietà A può essere scritto: A (PN (t), rN(t)) e fluttua nel tempo come risultato dell’interazione fra le particelle Il valore sperimentale della proprieta è la media di A nel tempo di misura: E’ quindi necessario conoscere come variano posizioni e velocità di ogni particella nel tempo, cosa che è possibile in linea di principio ma impossibile in pratica per un sistema macroscopico con 1023 particelle Meccanica statistica
Per risolvere questo problema è stata sviluppata la meccanica statistica in cui un singolo sistema che si evolve nel tempo è sostituito da un gran numero di repliche del sistema, considerate però in uno stesso istante (insieme) La media temporale è quindi sostituita dalla media sull’insieme: in cui l’integrale è esteso a tutte le coordinate e i momenti del sistema considerato, cioè d= dPNdrN Il sistema più comunemente usato in meccanica statistica é quello canonico in cui il numero di particelle N, il volume V e la temperatura T sono costanti In meccanica statistica si introduce poi il concetto di spazio delle fasi che altro non è un’estensione dello spazio conformazionale da una a più molecole
Un sistema di N-particelle è completamente definito dalle 3N coordinate e dai 3N momenti P=mv Un insieme di questi 6N valori (PN , rN)=(Px1, Py1,.., Pz3N ; x1, y1.., z3N) definisce uno stato del sistema o un punto nello spazio delle fasi. Ciascuna proprietà sperimentalmente misurabile A è ottenuta come media pesata di quella proprietà sullo spazio delle fasi: La funzione peso da la probabilità di trovare questo particolare stato del sistema nello spazio delle fasi, cioè la probabilità che il sistema abbia coordinate rN e momenti PN
Per l’insieme canonico (N V T costanti) la probabilità è data dall’espressione di Boltzmann function: • A (PN , rN) è il valore della proprietà A che il sistema assume nello stato (PN , rN) cioè in quel punto delle spazio delle fasi. • Ad esempio l’energia si ha: • E(PN , rN) = i=1,N Pi2/2mi + V(rN) d
In meccanica statistica il valore di una proprietà A è quindi ottenuto come media sull’insieme <A>, per calcolare la quale dobbiamo conoscere la probabilita che il sistema sia in un certo stato (PN , rN) Alternativamente possiamo propagare un singolo sistema nello spazio delle fasi e ottenere il valore di A come media temporale: L’ ipotesi ergodica stabilisce che: o meglio, visto che di necessita dobbiamo usare un tempo finito: Ipotesi Ergodica
Dinamica Molecolare Deterministica Monte Carlo Stocastica Propagazione del sistema nel tempo In tutte le tecniche di simulazione molecolare si genera una successione nel tempo di M configurazioni del sistema e per ciascuna si calcola A (PN , rN)
Energia Interna: Capacità termica Pressione: Temperatura: Proprietà Termodinamiche
Scelta del modello dell’energia Meccanica molecolare Ab initio Semiempirico Scelta del modello di confine Scelta della configurazione iniziale Fase di equilibrazione Dalla configurazione iniziale all’equilibrio Monitoraggio delle proprietà del sistema Fase di produzione Analisi della simulazione Impostazioni comuni della simulazione
Rimuove gli effetti di confine Permette il calcolo di proprie-tà macroscopiche usando un piccolo numero di particelle Quando una particella esce da una cella è sostituita dalla sua immagine che entra dalla cella sul lato opposto Condizioni al contorno periodiche
La successione di stati del sistema che si ottiene in una simulazione molecolare definisce un campionamento dello spazio delle fasi • Per ottenere buoni risultati il campionamento deve essere omogeneo e permettere di esplorare un’ampia parte dello spazio delle fasi • In particolare il campionamento è convergiuto se: • le quantità medie rimangono stabili nel tempo • Simulazioni da diverse condizioni iniziali danno gli stessi risultati Il problema del campionamento
Il metodo Monte Carlo genera casualmente delle configurazioni e usa degli particolari criteri per decidere se accettare o no ciascuna nuova configurazione nel set usato per il calcolo della proprietà A Questi criteri assicurano che la probabilità di ottenere una nuova configurazione sia uguale al fattore di Boltzmann Configurazioni di bassa energia vengono quindi generati con probabilità più alta che configurazioni di alta energia Alla fine dellla simulazione il valor medio della proprietà e ottenuto come semplice media
Nel metodo Monte Carlo l’energia è determinata solo dalla posizione degli atomi e non dai loro momenti E(PN , rN) = V(rN) La limitazione dovuta a questo fatto è che non si calcolano i valori assoluti delle proprietà ma solo le deviazione rispetto al valore per il gas ideale (facilmente calcolabile analiticamente) Non c’è alcuna relazione temporale fra configurazioni successive ottenute in una simulazione Monte Carlo
Integrazione Monte Carlo n. di punti sotto la curva _________________________________ Area sotto la curva = n. di punti totali
Energia potenziale media: Usando l’integrazione Monte Carlo Si generano Ntrial configurazioni casuali e si calcola: Proprietà termodinamiche Z
Questo approccio non è però fattibile per l’alto numero di configurazioni con fattore di Boltzmann molto piccolo Questo problema è ovviato nell’approccio Metropolis in cui le configurazioni sono generate con probabilità uguale proprio al fattore di Boltzmann L’integrale per Q o per una qualsiasi proprietà A può quindi essere approsimato da una semplice somma
Metropolis Monte Carlo Si generano selettivamente configurazioni che danno contributi maggiori all’ integrale Movimento casuale “Prova” DE Metropolis Test NO DE < 0 or exp(-DE/RT) < X[0,1] ? YES X[0,1] è un numero casuale fra 0 e 1
Simulazioni atomiche (esempio, cluster atomici) Simulazioni di liquidi Determinazione delle conformazioni molecolari (sistema di una sola molecola) Polimeri Applicazioni
Sistema di tre atomi uguali in una cella cubica Calcolo dell’energia potenziale totale media del sistema Esempio 1) Configurazione iniziale Energia potenziale E1: 2) Variazione casuale della posizione di un atomo qualsiasi, ad esempio l’atomo 2
3) Nuova configurazione Energia potenziale nuova configurazione E2: 4) Confronto energia configurazione precedente: a) Se E < 0 o exp(- E/RT) < X [0,1] la nuova configurazione è accettata b) Se exp(- E/RT) > X [0,1] la nuova configurazione non è accettata 5) Nel caso a) si riparte con una variazione casuale della precedente configurazione, la prima Nel caso b) si effettua una variazione casuale dell’ultima configurazione, la seconda
6) Si valuta l’energia di questa terza configurazione e si prosegue nello stesso modo 7) Si memorizzano le energie potenziali di tutte le configurazioni accettate E1, E2, E3,….. Dopo un certo numero di step, ad esempio 100, si conclude la simulazione e si calcola il valor medio dell’energia potenziale come semplice sommatoria:
A differenza del metodo Monte Carlo, nell’approccio della dinamica molecolare si calcola la dinamica reale del sistema Applicando le equazioni del moto di Newton si calcolano le traiettorie di tutte le N particelle del sistema La traiettoria di una particella specifica la posizione e la velocità di tale particella in funzione del tempo xi(t), yi(t), zi(t) vxi(t), vyi(t), vzi(t) ; basta conoscere le coordinate perché le velocità sono le derivate vxi(t) = dxi(t)/dt Data che la soluzione delle equazioni di Newton è numerica si ottiene una successione di posizioni e velocita in M istanti di tempo successivi t1, t2,…,tM : xi(t1), yi(t1), zi(t1) ; xi(t2), yi(t2), zi(t2) ; ………..; xi(tM), yi(tM), zi(tM) In pratica si ottiene una successione di M configurazioni che costituiscono un campionamento dello spazio delle fasi di tutto il sistema (N particelle)
Il valor medio di una proprietà A è ottenuta come media temporale in pratica in cui A(PN rN) = A( PN(ti ),rN(ti ) ) è il valore istantaneo della proprietà A che dipende dalle posizioni e dai momenti (velocità) delle N particelle nell’istante ti Si noti che a differenza del metodo Monte Carlo è necessario conoscere le velocità di tutte le particelle e non solo le posizioni
Propagazione temporale Equazioni del moto di Newton: Fi(r) = V(r1,r2,..,rN) / ri
Integrazione Numerica Espansione in serie di Taylor f(t+t) = f(t) + (df(t)/dt)t + 1/2 (d2f(t)/dt2)t2 + .. Per il moto di una particella ci interessa la sua posizione r e si ha: In cui v(t) =r(t)/dt e a(t)= d2r(t)/dt2e b(t) è la derivata terza
Algoritmo in pratica Si specificano le posizioni a t-dt e t. Dalle positioni si calcolano le forze al tempo t Si calcolano le posizioni al tempo t+ dt. Si calcolano le velocità Algoritmo di Verlet
Calcolo della forza al tempo t Energia di interazione fra gli atomi i e j Lennard-Jones Forza sull’atomo i dovuta all’atomo j
Calcola le forza dalla curvatura del potenziale Calcola le velocità dalla forza DT typically 10-15sec Calcola la nuova posizione Salva energia e geometria per la media Dinamica Molecolare
Per un sistema di atomi l’intervallo di tempo deve essere piccolo rispetto al tempo medio fra le collisioni Per molecole flessibili l’ intervallo ottimale è approssimativamente un decimo del tempo per il movimento più veloce Per molecole flessibili con legami flessibili il moto più veloce è quello di vibrazione, ad esempio per i legami C-H ~10 fs Gli algoritmi SHAKE e RATTLE sono usati per bloccare i gradi di libertà interni e permettere intervalli di tempo più lunghi Sistema Tipo di moto presente Intervallo suggerito Atomi Traslazione 10-14 Molecole rigide Traslazione, rotazione 5x10-15 Molecole flessibili Legami rigidi Traslazione, rotazione torsione 2x10-15 Molecole flessibili Legami flessibili Traslazione, rotazione Torsione, vibrazione 10-15 or 5x10-16 Scelta dell’intervallo di tempo
Definizione configuraz. iniziale Derivata sperimentalmente Calcolata Assegnazione delle velocità Inizio della simulazione Fase di equilibrazione Controllo della temperatura Fase di produzione Acquisizione dei dati Preparazione e avvio di una simulazione
Le componenti x, y, z delle velocità hanno distribuzione Velocità Le velocità sono assegnate casualmente da una distribuzione di Maxwell-Boltzmann alla temperatura di interesse:
Le velocità sono legate all’energia cinetica da: La temperatura è legata alle velocità da: Energia Cinetica e Temperatura
Dinamica e insiemi statistici Una simulazione dinamica è in genere eseguita a N, V e E costanti (insieme microcanonico) Può essere spesso utile eseguirla a N,V e T costanti (insieme canonico) Per eseguire una dinamica a T costante dobbiamo ad ogni step al tempo t calcolare T(t) e scalare le velocità, cioè moltiplicarle per un fattore , tale che la temperatura assuma il valore voluto T Infatti la temperatura è legata alle velocità da: Si dimostra che tale valore vale:
Sistema di tre atomi uguali in una cella cubica Calcolo dell’energia potenziale totale media del sistema Esempio 1) Configurazione iniziale (r1, r2, r2) al tempo t1 Energia interazione V12 V13 e V23 forze F12, F13,F23 ………. 2) Si calcola la nuova posizione al tempo t2=t1+t per ogni particella i=1,2,3
Solvatazione E importante poiché molte molecole sono in soluzione • Le molecole di solvente polare si dispongono attorno allo ione o alla molecola con il lato di polarità opposta rispetto alla carica (parziale) ed esercitano un campo elettrico • Questo campo elettrico da luogo ad un potenziale di interazione che deve essere aggiunto all’Hamiltoniano nell’equazione di Schroedinger • Nei metodi di meccanica molecolare si aggiunge semplicemente un contributo elettrostatico nell’espressione empirica dell’energia e si considera poi una costante dielettrica nell’interazione elettrostatica tra i vari atomi