Download

1 / 16
170 likes | 286 Views
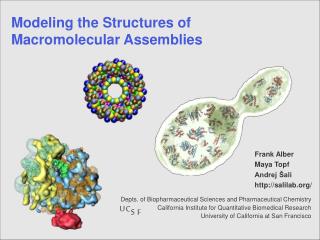
U. C. S. F. Modeling and Determining the Structures of Proteins and Macromolecular Assemblies. Depts. of Biopharmaceutical Sciences and Pharmaceutical Chemistry California Institute for Quantitative Biomedical Research University of California at San Francisco. Andrej Sali

E N D
U C S F Modeling and Determining the Structures of Proteins and Macromolecular Assemblies Depts. of Biopharmaceutical Sciences and Pharmaceutical Chemistry California Institute for Quantitative Biomedical Research University of California at San Francisco Andrej Sali http://salilab.org/
Protein structure models can be useful, despite errors D. Baker & A. Sali. Science 294, 93, 2001.
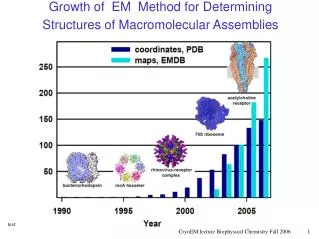
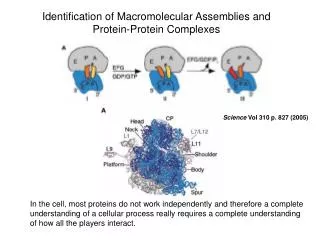
PHYSICS EXPERIMENT Determining the Structures of Proteins and Assemblies S Maximize efficiency, accuracy, resolution, and completeness of the structural coverage of proteins and their assemblies. Use structural information from any source: measurement, first principles, rules, resolution: low or high resolution to obtain the set of all models that are consistent with it. STATISTICS Sali, Earnest, Glaeser, Baumeister. From words to literature in structural proteomics. Nature 422, 216-225, 2003.
Characterizing Structures by Satisfaction of Spatial Restraints • Representation of a system. • Scoring function (spatial restraints). • Optimization. There is nothing but points and restraints on them.
p distance Scoring Function There is nothing but points and restraints on them. P (R / I) = ∏pi (ri / Ii ) i R … all degrees of freedom I … all information ri … ith restrained feature (eg, distance, angle, proximity, surface, density) Ii … information about ith restrained feature http://salilab.org/modeller/ Sali, Blundell. J. Mol. Biol. 234, 779, 1993. Alber, Kim, Sali. Structure 13, 435, 2005.
Hybrid modeling of the proteasome(Frank Alber) Best scoring model (out out 3,000 models) Native Starting structure Random configuration • Simulated from the known structure: • some subunit comparative models, • 3 crosslinks for some pairs, • 14 pullouts, • assembly shape (EM). drms ~1.3 nm
Spatial restraints on protein binary interactions from physics and bioinformatics
Identification of Binary Interactions by Structural Similarity Davis and Sali. Bioinformatics, 2005. Davis, Braberg, Shen, Pieper, Sali, Madhusudhan. Nucl. Acids Res., 2006.
Restraining binary protein interactions by computation • Comparative modeling • - based on structures of homologous complexes • - high accuracy • - low coverage (2) Protein docking - based on surface complementarity of target domains - high coverage - low accuracy (3) Comparative patch analysis - based on complementarity of target domains and binding sites of their homologs - higher accuracy than in docking and coverage than in comparative modeling
SH3 Structural characterization of PDZ3-SH3-GK fragment in rat PSD-95 PDZ2 PDZ3 PDZ1 GK PSD-95 domain architecture SH3-GK PDZ3 target subunits Results: predicted two alternate conformations (1) (2) • C-terminus binding cleft of PDZ3 and GMP binding site of GK become inaccessible upon binding • PDZ3 interacts with proline-rich binding site of SH3; • PXXP motif of PDZ3 might contribute to the interface • C-terminus binding cleft of PDZ3 is accessible
Experimental support of predictions • Limited proteolysis • - Limited proteolysis of recombinant PSD-95 was carried out using Proteinase K. • - A prominent ~48 kDa band at 30 minutes which corresponds to the PDZ3-SH3-GK fragment. • - Further digestion leads to appearance of a stable ~34 kDa band corresponding to the SH3-GK fragment. 2. Suggested experiments - Site-directed mutagenesis of the interface residues in the first proposed state could be used together with pull-down assays to validate the predicted interaction interface. - The lack of accessibility of the GMP-binding site in the second state could be tested using nucleotide-binding assays. - We expect the experimentally obtained SAXS spectra to be helpful in distinguishing the two PSD-95 states, based on the difference in theoretically predicted SAXS spectra.
Comparative patch analysis (Dmitry Korkin) Predicted complex Target domains
Data interpretation Translation into spatial restraints Modeling Modeling by satisfaction of spatial restraints Data generation Collection of experimental data Model interpretation Ensemble precision, accuracy The cycle of assembly structure characterization 3D structure embedding
Why Integrated Hierarchical System for Structural Biology? • No single method will give “best” possible assembly structure. • Need to integrate information to benefit from synergy among restraints. • Additional “information” provided by embedding in 3D. • Need to find all models consistent with the data, not just one. • Need to assess the results. • Provide feedback to guide future experiments. • “What if” simulations for optimized future experiments. • A way of “thinking” (information, “points of light” …).
Challenges • Robust, user friendly implementation. • More applications. • Expressing experimental information as restraints. • Combining restraints together. • Optimization. • Modeling of dynamic processes.
In Conclusion The goal is a comprehensive description of the multitude of interactions between molecular entities, which in turn is a prerequisite for the discovery of general structural principles that underlie all cellular processes. This goal will be achieved by a tight integration of experimental, physics-based, and statistics/rule-based approaches, spanning all relevant size and time scales. Sali, Earnest, Glaeser, Baumeister. From words to literature in structural proteomics. Nature 422, 216-225, 2003.