Absolute binding free energies
1.02k likes | 1.21k Views
...of ligands from umbrella sampling and steered MD simulations; ...applications to ions, small molecules and toxin peptides. Absolute binding free energies. Contents of this presentation I. Introduction. General principles of free energy calculations Illustrations by simple examples

Absolute binding free energies
E N D
Presentation Transcript
...of ligands from umbrella sampling and steered MD simulations; ...applications to ions, small molecules and toxin peptides. Absolute binding free energies
Contents of this presentation I Introduction • General principles of free energy calculations • Illustrations by simple examples • Toy models: ions in small water boxes • Theoretical Underpinnings • Ways to affirm and void the fidelity of simulation models to reality • Advanced examples • Ligands in gramicidin-A channel, K+ channels
Contents of this presentation II Introduction • Full analysis of an absolute free energy of binding calculation • Using toxin peptides (my work) • Summary and notes • Implications of various assumptions • Warning signs to watch out for ...let’s go!
Goal of FE calculations Introduction • Accuracy • Chief motivation of conducting calculations is to predict/replicate experimental values • We must not lose sight of this • Therefore, it is worthwhile to test and verify our foundations • Especially for calculations that last over a month
Mechanics of FE calculations Introduction • Umbrella sampling and steered MD are path-dependentcalculations e.g. the ligand must physically move out of the receptor • A well-constructed simulation can: • Take information from known reaction mechanisms • Provide information on how those mechanisms occure
Foundation of FE calculations Introduction F = ma • All simulations are models • Classical approximation of QM phenomena • System proceeds via forces imposed on atoms • Energy calculations occur via manipulation of the these forces • Small loss in accuracy must be granted ...motivated?
Steered molecular dynamics A simple manipulation F= ½ k ( r – r0 + tv ) • SMD is a “moving spring” • Force is imposed on an atom or set of atoms • Equilibrium position moves to create a path • Work done by this spring is used to find the local free energy surface.
A simple model Steered Molecular Dynamics • A small box, containing: • ~400 water atoms, • one neon atom. • Suppose that we create an artificial gaussian barrier (of ~5 kT) for the Ne atom. • How do I measure the free energy surface of that barrier, using the Ne as a probe?
Simulation design Steered Molecular Dynamics • Constrain position of Ne in (x,y,z) to a starting location • Define path along water box • Measure the total amount of work done W = ∫ F(z) dz • W → manipulations → free energy surface
Work done(?) Steered Molecular Dynamics • Resulting potential of mean force • Includes environmental influences • i.e. action of water around the atom • NB: notice that PMF does not return to zero readily
Averaging Steered Molecular Dynamics • A single trajectory does not “sample” the reaction sufficiently • i.e. A free energy surface requires comprehensive knowledge of the sub-states, ala possible trajectories • Trajectories contain influence from the SMD pulling itself • Some of the work is directed at displacing the solvent around Ne • This needs to be accounted for
Jarzynski’s Equality Steered Molecular Dynamics • .˙. Take multiple trajectories • The average of multiple trajectories should even out random influences on the PMF < e-W/kT > ≈ Σi=1ne-Wi / kT / n • .˙. Apply Jarzynski’s Equality (given certain assumptions) e-ΔG/kT = < e-W/kT >
Average work Steered Molecular Dynamics • The Boltzmann average of 10 calculations • v = 10 Å ns-1 • A very fast calculation, converges because Neon does not interact with environment ...next: theory
Criteria of SMD-based FE calculations SMD Assumptions • Reaction coordinate must be well chosen • This coordinate measures all contributions to the real ΔG, and only those contributions. • The simulation must be near-equilibrium • Jarzynski's Equality holds when no dissipative work is done by the pulling
Reaction coordinates SMD Assumptions In SMD, the dimension(s) controlled by the constraining potential is areaction coordinate • The reaction coordinate can be: • Distance • Center of mass (collective variable) • RMSD to target structure • The path traced through the SMD simulation is a reaction path
Theoretical interpretation SMD Assumptions • Our systems are canonical ensembles: Z(n,P,T) • We wish to measure particular sub-states of the system • e.g. ligand-bound and ligand-unbound. • Thus, steered molecular dynamics (SMD): • Constrains the system to certain sub-states according to some coordinate • Drives the system along this coordinate to new sub-states by application of forces.
Implications on path design SMD Assumptions • Both the reaction-path and the simulations must sufficiently sample the required sub-space • May not be complete, but must be representative • In a ligand-unbinding process • Translation must be primary • Rotation may be secondary Phase space Bulk Bound
Implications on path design SMD Assumptions • Sampling and stability of a system suffers where large barriers must be crossed • Therefore, choose path of least resistance Simply using the distance between the ligand and the whole receptor may not be a good choice…
Criteria of SMD-based FE calculations SMD Assumptions • Reaction coordinate must be well chosen • This coordinate measures all contributions to the real ΔG, and only those contributions. • The simulation must be conducted in a near-equilibrium state • Jarzynski's Equality holds when no dissipative work is done by the pulling
Theoretical implications SMD Assumptions SMD –(Jarzynski's Equality)→ Free energy • JE comes with certain qualifications: • Dissipative work can be done on the system • Pulling velocity i.e. System must equilibrate around SMD perturbation, else perturbation will also be measured. (We will show this later.) ...next:
Umbrella Sampling Collecting local information F= ½ k ( r – ri), i along path • US is a static potential • Force is also imposed on an atom or set of atoms • Multiple overlapping states are constructed to cover reaction path. • Each state provides information about local surface • Link to derive complete surface ...
Second toy model Umbrella sampling • A box containing two ions • sodium and chloride • Solution known • FE surface related to radial distribution function • This was done ab-initio yesterday
Second toy model Umbrella sampling • Reaction coordinate • Na – Cl separation • Umbrella potential • 1 Å apart, 2.5-9.5 Å • k = 10 kcal mol-1 Å-2 • Derive original by WHAM analysis
PMF convergence Umbrella sampling Phase space Bulk Bound
PMF Umbrella sampling Phase space Bulk Bound ...pretty...
Criteria of US-based FE calculations US assumptions • Reaction coordinate must be well chosen • This coordinate measures all contributions to the real ΔG, and only those contributions. • Sufficient sampling over the entire path: • Convergence of PMF curve means that environmental variables are well sampled. • Overlap between adjacent windows.
Reaction coordinate US assumptions • Same arguments as for SMD (underlying physics identical) • Umbrella sampling is capable of treating two/three dimensions • As long as all dimensions are properly sampled
Criteria of US-based FE calculations US assumptions • Reaction coordinate must be well chosen • This coordinate measures all contributions to the real ΔG, and only those contributions. • Sufficient sampling over the entire path: • Convergence of PMF curve means that environmental variables are well sampled • Overlap between adjacent windows
Environmental variables Bulk US assumptions • All coordinates not included in your reaction coordinate(s) must be well sampled • This means that simulation has visited dimensions perpendicular to reaction path that may contribute to FEbind Bound
Window Overlap Bulk US assumptions • Require enough sampling to accurately interpolate between windows Bound
Window-overlap US assumptions • Define measure of overlap between two distributions • When underlying surface is flat, harmonic potential produces gaussian distributions • Theoretical overlap: • Ω = [ 1- erf(d/8σ)]
Ω = [ 1- erf(d/8σ)] US assumptions • In practice, minimum overlap is ~2% • Overlap should agree with theoretical value when in bulkk = 20 kcal/mol/Å-2d = 0.5 ÅΩ = 15%k = 40 kcal/mol/Å-2d = 0.5 ÅΩ = 4%
Summary Steered MD –vs– Umbrella Sampling • Constructing FE surfaces via SMD • Straightforward construction • Relies on JE conditions: Difficult to achieve in practice • Constructing FE surfaces via US • Additional checks and balances • Both dependent on sufficient sampling ...take a break?
JE/US comparisons J Chem. Phys. 128:155104 (2008) • Using several testcases of ions andmolecules inchannel systems • Ion transit through membrane • Ion binding to gramicidin exterior • Organic-cation binding to gramicidin-A
JE/US comparisons J Chem. Phys. 128:155104 (2008) • Comparing PMFs • Tests balanced byequalising the totalsimulation time SMD setup @ v=5 Å ns-1 ~ US setup • SMD: Also use different pulling velocities to test reversibility of JE
Ion transit (nanotube) J Chem. Phys. 128:155104(2008) • Results equivalentbetween two methods • Energy surface not equal at both openings • resulting from system setup • JE valid
Ion transit (gA) J Chem. Phys. 128:155104(2008) • Umbrella sampling, not SMD, gives symmetric surface • Pulling at different velocities do not seem to help • Can potentially usev < 1 Å ns-1 • But more time consuming than equivalent US setup
JE: Practical problems? J Chem. Phys. 128:155104(2008) • Equilibration time sharply increases for peptide environments • Nanotube highly ordered .˙. Fast dissipation • Reliance on sampling “negative work” trajectories • High v: low probability for the environment to push SMD particle
K+-binding to gA J Chem. Phys. 128:155104(2008) ( next test case: ) • gA has a weak ion binding site at the entrances • Smaller potentials • Perhaps using a smaller k will reduce the perturbations v =2.5 Å ns-1
K+-binding to gA J Chem. Phys. 128:155104(2008) • What about using different force constants? • No significant help in repairing JE assumptions • Using small k may reduce perturbations on system • However, binding site shape lost • k must be greater than binding well ‘potential’ v =2.5 Å ns-1
K+-binding to gA J Chem. Phys. 128:155104(2008) • There are hard limits to varying parameters k = 2 kcal mol-1Å-2 k = 20 kcal mol-1Å-2
EA and TEA binding J Chem. Phys. 128:155104(2008) • Ethylammonium (EA) and tetra-ethylammonium (TEA) bind weakly to gA • v = 2.5 A /ns • k = 20 kcal mol-1 Å-2 • Reducing barrier height produces no difference here
CnErg1 Toy comparison J Chem. Phys. 128:155104(2008) • If it doesn’t work for small cations, it won’t work for a peptide • Test for a purported binding of CnERG1 toxin to hERG channel since rate of dissipation to environment is less than rate of work done… • SMD-PMFs essentially measures work done to move solvent
Mechanisms J Chem. Phys. 128:155104 (2008) • Input: work done on system • Carried out by imposed SMD forces (irreversible) • Contribution from underlying FE surface (reversible) • Output: dissipation to heat-bath (NPT systems) • Equilibration occurs by two means: • Forces bleed out to atoms far from SMD location • Temperature coupling to thermostat • JE maintained in O < I conditions (only in nanotube)
Mechanisms J Chem. Phys. 128:155104 (2008) • The time for equilibration issuch that v << 2.5 Ang/ns is required for JE condition to hold • This velocity requirement become more stringent as: • ligand size increases • interactions increase • Not as efficient as umbrella sampling. ...paper finished.
Organic cations-gA J. Phys. Chem. B 111:11303 (2007) • Block of GA by various small molecular ligands • Energetics dependent on ligand size and partial charges • Comparison between ligands, and with extant experimental data
Organic cations J. Phys. Chem. B 111:11303 (2007) • Use Autodock3 to find potential binding sites of molecules • MD, umbrella sampling to find PMF and free energy of binding • COM-coordinates of ligands 10 kcal mol-1Å-2 0.5 Å ns-1
Molecule list J. Phys. Chem. B 111:11303 (2007) • Use of six different molecules • Varying sizes and polarity • Determines strength of binding, and whether molecules can permeate through gA
MA and EA J. Phys. Chem. B 111:11303 (2007)
FMI and GNI J. Phys. Chem. B 111:11303 (2007)






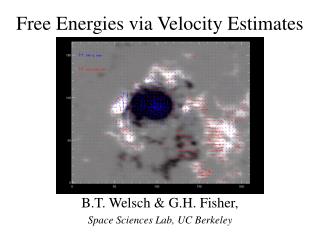
![Binding energies in DATA [MeV]](https://cdn2.slideserve.com/3921996/slide1-dt.jpg)