Download

1 / 14
150 likes | 424 Views
fenil Z etonuria. DEFINICIÓN. Hiperfenilalaninemia (HPA) ~ Fenilcetonuria (PKU). > 500 mutaciones. El defecto de la función a nivel hepático de la fenilalanina hidroxilasa (PAH) conlleva un aumento de la Phe en el organismo, que es el responsable de los efectos nocivos en el SNC:.

E N D
DEFINICIÓN • Hiperfenilalaninemia (HPA) ~ Fenilcetonuria (PKU) > 500 mutaciones El defecto de la función a nivel hepático de la fenilalanina hidroxilasa (PAH) conlleva un aumento de la Phe en el organismo, que es el responsable de los efectos nocivos en el SNC: La Phe compite con el transporte de Tyr (ya disminuida por síntesis) a través de la BHE y metabólicamente con la Gly, la Tyr, el Trp y el Glu cerebrales, además de otras inhibiciones enzimáticas (catalasa, glutatión peroxidasa, 3-OH-metilglutaril CoA reductasa…). También hay un acúmulo neurotóxico de metabolitos de la Phe, de los cuales el fenilacetato es el más tóxico. En el SNC se producen trastornos de la síntesis proteica, de la mielinización del cerebro, de la transmisión sináptica, de la síntesis de neurtransmisores (catecolaminas y serotonina), de la síntesis de colesterol, del estrés oxidativo y una disminución del metabolismo energético. Sistémicamente, se altera la síntesis de Tiroxina y Melanina, entre otras muchas proteínas. El déficit de síntesis/regeneración del cofactor BH4 (tetrahidrobiopterina) afecta a lahidroxilación de Phe, Tyr y Trp: Defecto de síntesis de catecolaminas, DOPA y serotonina (déficit de neurotransmisores) y daño en SNC, ello causa deterioro neurológico progresivo e incluso la muerte, por lo que se llaman HPA malignas o letales.
HISTORIA • La fenilcetonuria (PKU) (OMIM: 261600) es: • - La primera causa de conocida de retraso mental por trastorno metabólico. • - La primera enfermedad metabólica con tratamiento efectivo. Fue descrita por primera vez (1921) por la escritora Pearl S. Buck, en un relato sobre su propia hija. La identificación bioquímica comenzó en los años 20 (Noruega ), describiendo el olor especial de la orina (moho, ratón…). El Dr. Folling identificó que la orina se coloreaba de verde al añadir Cl3Fe. Posteriormente , identificó al ácido fenilpirúvico, como el causante de esta reacción y en 1934 describió la anormalidad bioquímica. El nombre de PKU fue atribuido al Dr. Penrose en la década de los años 30, siendo el primero que inició una dieta restrictiva, aunque sin éxito. En 1937 se identificó la transmisión genética, de forma autosómica recesiva, de la enfermedad y el Dr. Jervis identificó un defecto de la función de la enzima fenilalanina hidroxilasa como causante. El primer tratamiento dietético con éxito data de 1953 (Dr. Bickel) a base de un producto sustitutivo de proteínas libres de Phe. Actualmente se recomienda de por vida la dieta libre de Phe. El primer cribado neonatal se desarrolló al final de los años 50 midiendo la Phe en papel seco. En 1968 lo inició en Granada el Dr. Mayor Zaragoza, siendo actualmente universal en España. En los años 80 se localizó el gen PHA, en 12q24.1, del cromosoma 12.
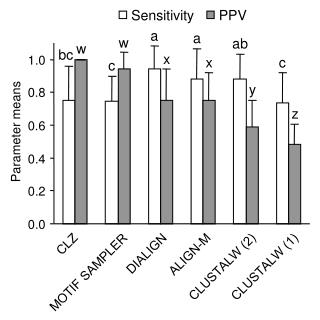
CRIBADO NEONATAL CRITERIOS DE INCLUSIÓN CRIBADO NEONATAL: - Elevada morbi-mortalidad si no se diagnostica en neonato: - Pasa inadvertida en examen físico: - Tratamiento efectivo: - Prevalencia > 1/10.000: - Cribado analítico eficiente: Cursa con retraso mental y motor grave, comportamiento psicótico, hiperactividad, déficit de atención, EEG anormal, convulsiones, hipopigmentación, olor característico (sudor y orina). Las formas menos graves cursan con baja concentración y mal rendimiento escolar. Sintomatología clínica hacia los 6 meses de edad, si no se trata desde el nacimiento. Restricción dietética de Phe, en mayor o menor grado, según la gravedad. El tratamiento con cofactor (BH4) puede mejorar la tolerancia a Phe. Es sencillo y barato, pero difícil en la práctica (seguimiento estricto). En el periodo 1988-2010 1:19336 para PKU y 1:11964 para HPA Phe en sangre impregnada en papel. Los métodos analíticos son variados: Cromatografía capa fina, espectrometría de masas en tándem (MS-MS), espectrofotometría y fluorimetría, en base a las cuales el punto de corte varía Phe = 2 - 2.5mg/dL y Phe/Tyr =2.5.
CRIBADO NEONATAL Extracción a partir del tercer día de vida, tras 48 horas de toma de alimento, aunque no existe un consenso universal. MS/MS - Separa y cuantifica iones basándose en la razón masa molecular/carga y presenta los resultados en un espectro donde cada molécula se identifica según su peso y concentración. - Detecta muchos errores congénitos del metabolismo en un único análisis clínico. - Proyecto Region-4-Genetics: 100 laboratorios / 5 continentes / Aas & Acil-carnitinas
CLASIFICACIÓN • HPA / PKU (autosómicas recesivas): • Defectos en el cofactor BH4 (autosómicas recesivas / HPA malignas): • Phe = 2.5 - 6 mg/dL • (Tolerancia de Phe en dieta > 600 mg/día). • Phe = 6 - 10 mg/dL. • (Tolerancia de Phe en dieta de 400 - 600 mg/día). • Phe = 10 - 20 mg/dL. • (Tolerancia de Phe en dieta de 350- 400 mg/día). • Phe > 30 mg/dL. • (Tolerancia de Phe en dieta < 350 mg/día). - HPA: - PKU leve: - PKU moderada: - PKU grave o clásica: 1 g de proteínas de alto valor biológico 50 mg de Phe. 1 mg/dL = 60 mmol/L. • Phe > 2.5 mg/dL • (GTP ciclohidrolasa, 6-Piruvoil-tetrahidropterin sintasa o Sepiapterina reductasa). • Phe > 2.5 mg/dL • (Pterina-4-carbinolamina deshidratasa o Dihidropterina reductasa). - En la síntesis : -En la regeneración:
FENOTIPO CLÍNICO - PKU clásica no tratada : - PKU clásica tratada: - HPA moderada o benigna: - HPA transitoria del recién nacido: - Embiopatía por PKU materna: - HPA maligna : Retraso mental y motor severo, epilepsia, hiperactividad, rasgos psicóticos (agresividad incontrolada y automutilaciones) y demencia. Rasgos físicos como microcefalia, ojos, piel y cabellos claros, olor corporal especial y eccema. Coeficiente de inteligencia en los límites de la normalidad, aunque presentan dificultades en el aprendizaje, torpeza motriz, dificultades grafoperceptivas, hiperactividad y trastornos del sueño . El déficit enzimático es parcial, no suele requerir tratamiento y la evolución es favorable, aunque pueden presentar alteraciones en la atención y en el sueño. En prematuros e hijos de madres PKU, suele ser transitoria y no deja secuelas. Embriopatía en hijos de madres con PKU grave/moderada, con niveles de Phe elevados en meses previos y/o gestación. Se evita manteniendo Phe =2-6 mg/dL desde 8 semanas previas a la gestación hasta el final de la misma Si no se controla, puede cursar con retraso mental y del crecimiento, malformaciones cardiacas, microcefalia y dismorfias faciales. Debida a alteraciones en la BH4. No responde al tratamiento. Existe deterioro neurológico por déficit de catecolaminas y serotonina. Además, presentan dificultad respiratoria, parkinsonismo, mioclonus, corea, signos piramidales, epilepsia rebelde…
DIAGNÓSTICO DIFERENCIAL ¿QUIÉN? Phe> 2.5 mg/dL en el Cribado neonatal ¿POR QUÉ? PKU/HPA ó déficit BH4(1) Estudio diversos analitos Eficacia tratamiento BH4(2) Sobrecarga con BH4 (1)¿CUÁNDO? RN: 3 días con 500-700 mg Phe/día (10-14 g PAVB/día) No RN: RN: 3 días con 150 mg Phe/Kg·día (g PAVB/Kg·día) (1)¿QUÉ y CÓMO? Plasma Análisis cuantitativo de Aas. Confirma Phe del cribado & Phe + Tyr + Aas. CII, HPLC, MS-MS. Sangre total Dihidroptrerina reductasa en eritrocitos.(papel S&S) Orina Confirma Phe del cribado & Metabolitos de Phe (oscuridad) (ács. mandélico, fenilpirúvico, fenil-láctico...) por MS-MS Neopterina y biopterina por HPLC. AHV y 5-OH-Indolacético (metabolitos de neurotransmisores) por HPLC. LCR Metabolitos de BH4, serotonina y catecolaminas (oscuridad) por HPLC. (2)SOBRECARGA Dosis en RN:BH4 20 mg/Kg. ORAL CON BH4 Sangre total(papel S&S): Basal, 2, 4, 6, 8, 10, 12, 16, 20 y 24 h. Orina: Basal, 0-8 h, 8-24 h.
DIAGNÓSTICO DIFERENCIAL (1) Clasificación según el diagnóstico diferencial: Deficiencia de Fenilalaninahidroxilasa (PAH): PKU: Phe > 6 mg/dLTyr< 2.1 mg/dL. HPA: Phe = 2.5-6 mg/dLTyr< 2.1 mg/dL Neopterina y Biopterina (%Biopterina = 20-80%). DHPR normal Deficiencia de GTP-ciclohidrolasa (GPT-CH): Phe > 2.5 mg/dLTyr <2.1 mg/dL. Neopterina y Biopterina (%Biopterina = 20-80%). DHPR normal Deficiencia de 6-piruvoil-tetrahidro-biopterina-sintetasa (PTPS): Phe > 2.5 mg/dLTyr < 2.1 mg/dL. Neopterina y Biopterina (%Biopterina < 19%). DHPR normal Deficiencia de Dihidrobiopterinareductasa(DHPR): Phe > 2.5 mg/dLTyr < 2.1 mg/dL. Neopterina normal y Biopterina (%Biopterina > 83%). DHPR Deficiencia de carbinolaminadehidratasa (PCD): Phe > 2.5 mg/dLTyr <2.1 mg/dL. Presencia de Primapterina. DHPR normal.
DIAGNÓSTICO DIFERENCIAL (2) Evaluación de la S.O.BH4 para la eficacia del tratamiento: Deficiencia de Fenilalaninahidroxilasa (PAH): PKU sensible: Phe > 29% a las 8h post-S.O.BH4, respecto a Phebasal. PKU respondedora lenta: Phe > 29% a las 12-16h post-S.O.BH4, respecto a Phebasal. Deficiencia de GTP-ciclohidrolasa (GPT-CH): Phe < 2.5 mg/dL a las 4 h de S.O.BH4. Biopterina desde las 4h post-S.O.BH4. Deficiencia de 6-piruvoil-tetrahidro-biopterina-sintetasa (PTPS): Phe <2.5 mg/dL a las 4 h de S.O.BH4. Biopterina desde las 4h post-S.O.BH4 Deficiencias de regeneración Dihidrobiopterinareductasa(DHPR) y de Carbinolaminadehidratasa (PCD): Phe no normaliza. No cambios en pterinas. Diagnóstico genético: >500 mutaciones conocidas en la PAH. Todos los genes correspondientes a los defectos de actividad de PAH, así como los de síntesis y regeneración de BH4, son de herencia autosómica recesiva.
TRATAMIENTO - El objetivo fundamental va a consistir en reducir las cifras de Phe en sangre lo suficiente para prevenir los efectos secundarios. • El único tratamiento eficaz es la restricción dietética de Phe, reduciendo el aporte de alimentos que contengan Phe y suplementándolo con fórmulas especiales libres de Phe pero con el resto de aas y nutrientes esenciales. • Todos los alimentos con alto valor biológico (carnes, pescados, huevos, leche y derivados) son los que más cantidad de Phe contienen. Otros como cereales y legumbres, son alimentos que estarán controlados. Mientras que verduras, hortalizas y frutas su consumo podrá ser mayor. - Hay que seguir unas pautas para un tratamiento sea óptimo: * Diagnóstico precoz. * Control riguroso durante el embarazo en madres fenilcetonúricas. * Tratamiento y control periódico durante la infancia, adolescencia y vida adulta: Phe < 6 mg/dL (infancia) Phe < 10 mg/dL (adulto). - Pacientes HPA Si presentan Phe < 6 mg/dL o Phe < 3 mg/dL en mujeres embarazadas afectas, sin restricción, no precisarán de un tratamiento especifico. - Fenilcetonuria sensible a BH4 El aporte extra de BH4 dependerá de la respuesta obtenida a la sobrecarga con BH4 de cada paciente, sobre todo en fenotipos de PAH muy sensibles a BH4. - Nuevas terapias: * Aas largos neutros Compiten con Phe por el transporte en la BHE. Reduce las concentración de Phe en cerebro y aumenta la síntesis de neurotransmisores a nivel cerebral. * Fenilalanina Amonio Liasa (PAL) Enzima que también degrada la Phe pero es más estable que PAH y no requiere la ayuda del cofactor.