Download
1 / 44
440 likes | 456 Views
Learn about the principles of NMR spectroscopy, including the role of hydrogen nuclei (protons), spin quantum numbers, magnetic moments, isotope characteristics, Larmor frequency, instrument frequencies, and the significance of magnetic fields in NMR experiments.

E N D
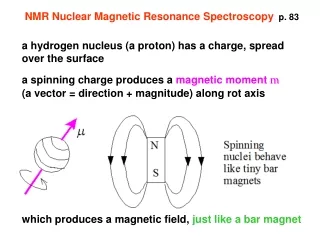
NMR Nuclear Magnetic Resonance Spectroscopyp. 83 a hydrogen nucleus (a proton) has a charge, spread over the surface a spinning charge produces a magnetic moment m (a vector = direction + magnitude) along rot axis which produces a magnetic field, just like a bar magnet
p. 83 this moment interacts with an external field, just like a magnet N B0 S BUT only certain orientations are allowed, which depends upon the nuclear spin quantum number I
p. 83 The magnetic moment, m is proportional to the spin quantum number I m = gI (h/2p) where g = Magnetogyric ratio = magnetic moment angular momentum • is different for each nucleus (a constant) I is allowed only certain values and these values can range from mI = –I to +I in integer steps ONLY Our proton is simple as it has I= ½, so only two values of mIare permitted, +1/2 and -1/2
In ‘bar magnet’ terms, these values of mI correspond to the magnet aligned with and opposed to the external field or B0 = mI = +1/2 -1/2 However, these two states ONLY have different energy in a non-zero external field, B0 E B0 DE = hn B0 = 0 p. 83/84
p. 84 B0 DE = hn so, if we put ‘our proton’ in a strong magnetic field, B0, there are TWO nuclear spin states: lower energy = aligned with field (mI = +1/2) higher energy = opposed to field (mI = -1/2) A transition will occur at a frequency n this is in the radiofrequency range,~108Hz (100 MHz) and this is the basis of the NMR (or MRI) experiment we make a nucleus‘flip’its spin
However, we do NOT really have little bar magnets!!! p. 84 But this frequency, n is the same as what we have called the ‘flip frequency’ NOTE:n is proportional to B0 – more about that later
p. 85 The spin number, I depends on the isotope x = number of protons + neutrons = mass x E Y = number of protons = charge = Atomic # Y e.g. 11H has mass of 1 and charge of 1 21H = D has mass 2, charge 1 mass 2 = 1 proton + 1 neutron IF x and y are both even, I = 0, NOT NMR ACTIVE e.g. 126C 168O 42He Common isotopes
p. 85 If charge (y) is odd and mass (x) is even, I = integer e.g. 21D and 147N both have I = 1 If mass is odd, I = Integer/2 11H, 136C199F 3115P all have I = 1/2 IMPORTANT 3517Cl I = 3/2; 178O I = 5/2 too difficult for now
Number of spin states p. 85 Nuclei have spin states of magnetic quantum number MI = +I, (I-1), (I-2)...-I where total number of states is then 2I+1 so if I = ½ , # states = 2, and MI = +1/2 and -1/2 if I = 1, # = 3, MI = +1, 0, -1 case for Deuterium if I = 3/2, # = 4, MI = +3/2, +1/2, -1/2, -3/2 case for 11B
p. 86 I = ½ 1H I = 3/2 11B
p. 86 E B0 DE = hn B0 = 0 This frequency is the Larmor frequency, where: gB0 n = 2p so irradiation of nucleus with frequency n causes a transition from lower to upper state which we call ‘flipping the spin’ This frequency depends on B0 (field) and g (nucleus type)
p. 86/87 gB0 Earth’s magnetic field is ca. 0.5 G at surface n = 2p For 1H, g = 267.51 (radian MHz per Tesla) Then when B0 = 2.35 Tesla, n = 100MHz [1 Tesla = 10,000 gauss = 10kGauss] so for each Tesla of B0, n = 42.55 MHz so for each 100 MHz you need 2.35 Tesla of B0 Our largest departmental NMR is referred to as a 500 MHz instrument because this is the resonance frequency of 1H at its magnetic field of 11.74 Tesla(cost ~ 500K$)
p. 86 Magnetogyric ratios nucleus ggrel (relative freq to 1H) 1H 267.1 1 19F 251.8 0.94 31P 108.40 0.40 11B 85.84 0.32 13C 67.28 0.25 29Si 53.15 0.19 2H 41.06 0.15 n, MHz
p. 87 For different nuclei: n1 = k g1 B1n2 = k g2 B2 k = 1/2p n1g1 B1 = so n2g2 B2 So if know five of these we can calculate other but note that B1 = B2 for the same instrument
p. 87 NMR Instruments Known by what PROTON frequency they operate at e.g. we have 300, 360 and 500 MHz instruments these all can run carbon spectra, and since grel for 13C = 0.25, these same instruments operate at 75, 90 and 125MHz respectively when running carbon spectra [magnetic field strength is essentially fixed for each instrument] If all protons absorbed at exactly the same frequency n the NMR experiment would NOT be very useful! gB0 n = 2p
Bactual = B0 + Blocal p. 87 Fortunately, this is not the case: electrons have an associated magnetic field too so small differences in the electronic environment can cause one 1H nucleus to resonate at a slightly different n than another gB0 alcohol n = 2p Essentially, the applied field is slightly modified by the local electronic environment: n is a very sensitive probe of the local field B and we can measure radiofrequencies very accurately
p. 88 SHIELDING: local electronic effects on Bnet e- Bo Binduced Electrons in bonds circulate in a magnetic field creating an opposing magnetic field Binducedthat cancels part of Bo More e- means a bigger Binduced and a smaller Bnet so to reach resonance, you would need to apply a higher Bo (or, if Bo is fixed, a lower ν is required)
p. 88/89 CHEMICAL SHIFT, d Variations in ncaused by local effects are on the order of 1 part in 105 to 107 of the ‘normal’1H resonant n We COULD just quote the absolute resonant frequencies • for a given 1H type BUT: • The differences would be tiny and unwieldy to quote: e.g. 250.000000 vs. 250.000002 MHz b) Absolute frequencies vary linearly with B0: e.g. 250.000002 vs. 500.000004 on a 250 vs. 500 MHz machine
p. 88 CHEMICAL SHIFT, d A better way: measure all resonances RELATIVE to a standard and divide by B0(or more simply the standard resonance frequency for a given nucleus at a particular B0) to remove the field dependence The Standard: TMS = tetramethylsilane = Si(CH3)4 = 0 Sample peak n - TMS peak n Hz = ppm d = = Spectrometer n in MHz MHz shift in Hz n TMS sample
p. 89 CHCl3 = 7.2 ppm The shift from TMS is thus 7.2 x spectrometer operating frequency in MHz e.g. at 100MHz = 7.2 x 100 = 720 Hz at 300MHz = 7.2 x 300 = 2160 Hz at 500MHz = 7.2 x 500 = 3600 Hz Thus the absolute shift depends upon the instrument, but the chemical shift does not (7.2 ppm) Higher frequency (larger field) instruments are more sensitive, and more expensive!
SUMMARY Nuclei have spin I (depends on # protons/neutrons) Spinning charges = magnetic field Nuclear spin I quantized: only mI states from +I,..,-I allowed, each with a different magnetic moment mI states same energy unless placed in an external magnetic field: + mI (spin aligned with ext. field) lower energy than - mI NMR experiment probes nrequired to ‘flip’ spin: cause nuclear spin state to change (eg. mI = 1/2 to -1/2) n depends on ext. field strength but also on tiny variations in electron density (because e- are spinning charges too) in the locale of the nucleus: we can tell something about the nuclear environment from n
p. 90 Deshielded Downfield Low field TMS 5.7 3.5 7.3 3.7
= integration = area under peak is proportional to NUMBER of H’s = 1:3 p. 90 d 7.3 3.7 it is a RATIO, i.e 1:3 or 2:6 or 3:9 etc
= integration = area under peak is proportional to NUMBER of H’s = 2:3 p. 90 TMS it is a RATIO, i.e 2:3 or 4:6 or 6:9 etc
p. 91 d 1 d 3.5 size ratio 3:1
p. 92 SPIN-SPIN COUPLING F---PCl2 Both P and F have spin I = ½ (we can ignore Cl here) high n low fieldhigh field low n
p. 93 THE COUPLING CONSTANT J nJXY where n = number of bonds between coupled the nuclei X = nucleus that is being observed Y = nucleus that is coupling to X (often the order of XY or YX is arbitrary) 4JFH 2JPF 3JHH
p. 93 d = chemical shift = centre of all lines (ppm) J = coupling constant = separation of lines (Hz) s, d, t, q = quartet, pentet, sextet, septet, octet nonet, decet
p. 93 ** HA=HB HA HB **cannot see coupling between identical nuclei
More nuclei – AX2 e.g. PF2Cl The P can see both fluorines up (twice as likely) one up, one down both down so there are THREE lines, the middle one has twice the intensity
p. 94 TREE DIAGRAMS AX2 A A split by X JAX split by X again J/2 J/2 J/2 J/2 Can always construct pattern as a tree, by splitting one nucleus at a time – when nuclei are same, e.g. X above, lines fall on top of each other and simplify pattern
p. 94 AX AX2 AX3 AXn AX4 Spectrum of A: (2nI + 1) lines 4+1 = 5 lines I = spin of X; n = number of X atoms For I = ½ , = n + 1 lines 1:4:6:4:1 intensities
p. 94 Numerically, we can get intensities from Pascal’s triangle: 1 1 1 1 2 1 1 3 3 1 1 4 6 4 1 1 5 10 10 5 1 1+5 =6 5+10 =15 10+10 =20 5+1 =6 1 10+5 =15 1 so, intensities of a septet are 1:6:15:20:15:6:1
Example A p. 95 3 neighbors, so 3+1=4 lines 2 neighbors, so 2+1 = 3 lines 1:3:3:1 Br----CH2-----CH3 1:2:1
Example B p. 95 CHCl2—CHCl---CHCl2 2 identical neighbors, so triplet 1 neighbor, so doublet
Example C p. 96 If J’s are different, best to draw a tree, e.g. PHFCl You need to know J’s, assume 1JPF > 1JPH for now P In 31P Spectrum: 1JPF P split by F 1JPH 1JPH split again by H d so spectrum is a doublet of doublets
Example D p. 96 PH2F P Psplit by F split again by 2H get a doublet of triplets
p. 96 For AXYZ draw tree by starting with largest J then next largest J then smallest J then lines will cross over each other the least! Consider CHF2CH3 H is triplet of quartets H2is doublet 13CF3CH2CH3 of quartets of quartets
SIZE OF COUPLING CONSTANTS Table, manual p. 97 Generally 1J >> 2J >> 3J > 4J > 5J > 6J Ball park figures!! 200Hz50Hz10Hz 3Hz 1Hz but there are plenty of exceptions >CHAHB ~15 =CHAHB~2 * only P, F,.... 1 Hz 0 Hz * For H-H = 0, except in pi-bonds 10-18Hz 7Hz
Example E p. 98 C5H8F4O C5H8F4O - C5H12O = 0 DBE Integration = 1:1:6 ---8H total Peak at d 6 is 1H t of t (triplet of triplets)
p. 98 C5H8F4O Integration = 1:1:6 ---8H total so t of t must be caused by F’s arranged 2 + 2 Other H signals are singlets There is a large coupling and a small coupling Large Coupling ~ 1ppm =60Hz = 2J = H-C-F2 small coupling ~0.1ppm =6Hz = 3J = H-C-C-F2 so we have CHF2-CF2- group not split by anything else
p. 98 C5H8F4O Integration = 1:1:6 ---8H total CHF2—CF2-- 6H’s are identical and are NOT split by neighbors we have 3C’s, O, 7H’s to find X, Y not H or F otherwise would see coupling 3J X—C(CH3)2 Y One of these must be CHF2CF2
p. 98 C5H8F4O CHF2—CF2-- Integration = 1:1:6 ---8H total C—C(CH3)2 now groups left to attach are H and CHF2CF2-- O so CHF2CF2—C(CH3)2 we will learn later that OH usually do not couple OH
You can now do ASSIGNMENT 4